
Adult-onset hemophagocytic syndrome with severe central nervous system involvement: a case report
Zan Wang
Affiliation
Department of Neurology, The First Hospital of Jilin University, Changchun, China
Corresponding Author
Zan Wang, MD, PhD, Professor, Department of Neurology, the First Hospital of Jilin University, Changchun 130021, China, fax: (+86 0431) 88782378, Tel: (+86 0431) 88782378; E-mail: wangzanprof@163.com
Citation
Wang, Z., et al. Adult-Onset Haemophagocytic Syndromewith Severe Central Nervous System Involvement: A Case Report.(2018) Int J Hemat Ther 4(2): 53- 57.
Copy rights
© 2018 Wang, Z. This is an Open access article distributed underthe terms of Creative Commons Attribution 4.0 International License.
Keywords
Haemophagocytic syndrome; Central nervous system; Pathology; Infect
Abstract
This study reports a case of adult Haemo Phagocytic Syndrome (HPS) with severe central nervous system involvement.It is easily misdiagnosed because there is no specificity in the imaging findings. This should be considered in the differentialdiagnosis of adults presenting with progressive brain lesions, even in the absence of typical systemic signs ofHPS. A bone marrow biopsy is the key to an early diagnosis.
Introduction
Haemo Phagocytic Syndrome (HPS), also known as Haemophagocytic Lympho Histiocytosis (HLH), is caused by a variety of factors in macrophages and the abnormal activation of large quantities of phagocytic blood cells[1-3]. It can cause serious and even fatal excessive inflammatory response syndrome, including:[4] persistent fever, hepatosplenomegaly, pancytopenia, coagulation disorders, abnormal liver function, hypofibrinogenaemia, and high glycerine trilaurate, among other conditions. HPS is a typical systemic disease with variable degrees of Central Nervous System (CNS) involvement, which can occur with meningitis, seizures, and psychiatric symptoms[5,6]. Leukaemia may also rarely present with CNS involvement and unusual symptoms, Piccin A et al[7] reported two case, which showed hypophyseal damage in erythroleukaemia with diabetes insipidus relating to specific chromosomal/molecular alterations. HPS is subdivided into primary and secondary haemophagocytic syndrome. Primary HLH mainly includes Familial Hemophagocytic Lympho Histiocytosis (FHLH) and immunodeficiency syndrome associated HLH. FHLH has a clear family genetic or genetic defect, 80% FHLH patients develop the syndromes in infants, but very rare in adulthood. A later onset of the disease has been sporadically reported in adolescents and adults. Sieni E et al[8] reported their retrospective analysis results of all FHLH cases at an age of 18 years or older and enrolled in the Italian Registry of HLH. A total of 11 patients were diagnosed with FHLH based on the evidence of genetic defect in one FHLH-related gene, their age ranged from 18 to 43 years (median, 23 years). Secondary haemophagocytic syndrome have been linked to acquired viral infections, malignancies, prolonged intravenous nutrition, organ/bone marrow transplantation, and autoimmune disorders[2]. The majority of cases of Central Nervous System HPS (CNS-HPS) occur in the paediatric population
This report presents a rare case of an adult-onset patient with HPS and concurrent CNS involvement, which provides the experience for the diagnosis of adult-onset CNS- HPS.
In May 2016, a 19-year-old male university student was referred to our hospital complaining of persistent fever for 1 month, seizures for 10 days, and unconsciousness for 1 day. One month before admission, he began to have a fever, with the highest temperature up to 39.2°C and fatigue. He was treated with antibiotics (detailed information was not provided) in a local hospital, but the fever persisted. He continued to be febrile and developed weakness and generalised seizure. A brain MRI showed multiple abnormal signals in the bilateral frontal lobe, occipital lobe, parietal cortex, and subcortical white matter. The patient was suspected of having an intracranial infection. However, a routine blood examination showed pancytopenia. The results of bone marrow puncture of the patients showed haemophagocytosis. He was thought to have HPS. His condition gradually worsened, and he lost consciousness. He was then sent to our hospital. On admission, a neurologic examination revealed a symmetric proximal tetra paresis (3/5 on the MRC scale), increased muscle tension, and negative bilateral Babinski’s sign.
The patient’s laboratory examinations were as follows
(Table 1): Abdomen ultrasound examination revealed mild hepatosplenomegaly and seroperitoneum. EEG for eight hours: the background EEG amplitude regulation was poor, and there were no typical epileptiform discharges during wakefulness and focal slow activity. Lung CT showed pneumonia with pleural effusion. Brain MRI (on May 28, 2016) of the bilateral frontal lobe, occipital lobe, and parietal cortex revealed multiple abnormal signals (Figure 1). Brain MRI (on June 6, 2016) showed the bilateral parietal lobe and right temporal occipital multiple abnormal signals (Figure 2). Brain MRI spectrum analysis (on June 17, 2016) demonstrated the MRS change of the right occipital lobe, indicating the possibility of non-neoplastic lesions (Figure 3).
| Laboratory examination | Result | Normal range |
|---|---|---|
| White blood cell (WBC) | 1.78 X 10^9/l | 3.5 - 9.5 X 10^9/l |
| hemoglobin (Hb) | 82 g/l | 130 - 175 g/l |
| blood platelet(PLT) | 79 X 10^9/l | 125 - 350 X 10^9/l |
| liver function | ||
| AST | 250.8 IU/L | 9 - 50 IU/L |
| ALT | 503.3 IU/L | 15 - 40 IU/L |
| gamma glutamyltransferase (GGT) | 162.4 u/l | 10 - 60 IU/L |
| Total protein (TP ) | 55.4 g/l | 65 - 85 g/l |
| albumin (Alb ) | 26.5 G/l | 40 – 55 g/l |
| Triglyceride (TG ) | 3.13 mmol/l | < 1.7 mmol/l |
| fibrinogen | 1.43 g/l | 2 - 4 g/l |
| Ferritin | 555.21 ng/ml | 21.8 - 274.6 ng/ml |
| torch virus antibody (serum) | ||
| EB virus antibody IgG | (+) | (-) |
| cytomegalovirus antibody IgG | (+) | (-) |
| cytomegalovirus antibody IgM | (+) | (-) |
| herpes simplex virus antibody IgG | (+) | (-) |
| rubella virus antibody IgG | (+) | (-) |
| Tumor markers: | ||
| carcinoembryonic antigen(CEA) | 6.61 ng/ml | < 5 ng/ml |
| carbohydrate antigen 125 (CA125) | 93.27 u/ml | < 35 u/ml |
| Cerebrospinal fluid (CSF) | ||
| WBC | 5 * 10^6/l | 0 - 8 X10^6/l |
| Protein | 0.58 g/l | 0.15 - 0.45g/l |
| sugar | 3.24 mmol/l | 2.3 - 4.1 mmol/l |
| chlorine | 127 mmol/l | 119 - 129 mmol/l |
| Pan’s reaction | (+) | (-) |
| Blood Soluble interleukin-2 receptor(sil-2r) | 786 u/ml | 223-710 u/ml |
| NK cell (cd3-cd56+) | 2% | 8.75 - 40.52% |
| CMV-PP65 | (+) | (-) |
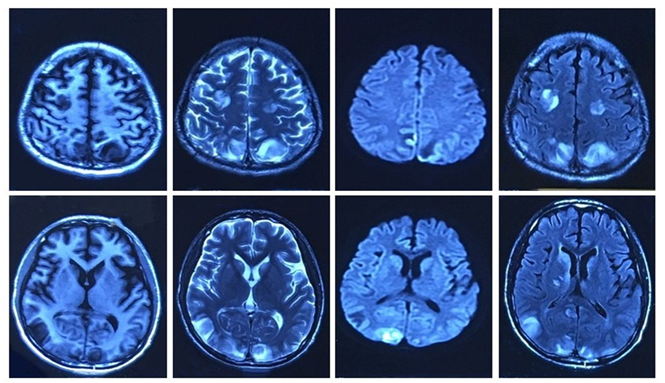
Figure 1: brain MRI (2016.05.28): The bilateral frontal lobe, occipital lobe and parietal lobe subcortical show slightly long T1 signal and long T2 signal, these signals are inhomogeneous. The DWI phase and the flair phase present hyperintense.
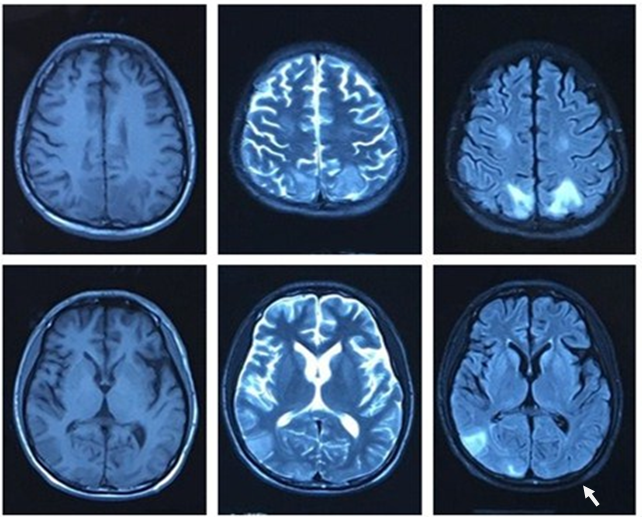
Figure 2: brain MRI (2016.06.06): The bilateral parietal lobe, right temporal lobe and occipital lobe show slightly long T1 signal and long T2 signal. The flair phase present hyperintense.
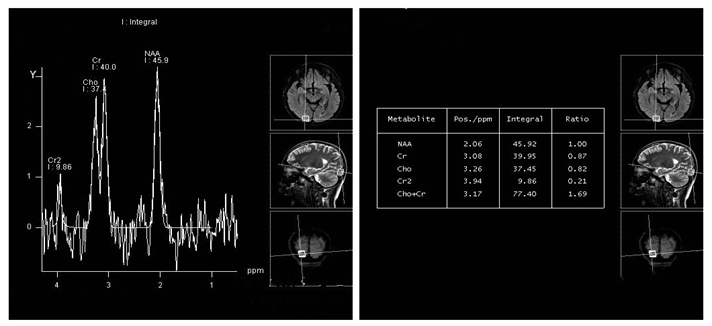
Figure 3: brain MRI spectrumanalysis (2016.06.17): Flair :Right occipital lobe, bilateral parietal lobegyrus have patchy hyperintense. The right occipital region was selected: Cho, the Cr peak increased insignificantly, the value of Cho + Cr/NAA is about 1.69. The MRS change of right occipital lobe is considered non-neoplastic lesions.
Routine blood examination: White blood cells (WBC) 1.78 × 10^9/l (normal range: 3.5 - 9.5 × 10^9/l), haemoglobin (Hb) 82 g / l (normal range: 130 - 175 g / l), blood platelet (PLT) 79 × 10^9/ l (normal range: 125 - 350 × 10^9/l). Liver function: AST 250.8 IU/L (normal range: 9 - 50 IU/L), ALT 503.3I U/L (normal range: 15 - 40 IU / L), Gamma Glutamyl Transferase (GGT) 162.4 u / l (normal range: 10 - 60 IU / L). Total protein (TP) 55.4 g / l (normal range: 65 - 85 g / l), albumin (Alb) 26.5 G/l (normal range: 40 - 55 g / l). Tri Glyceride (TG) 3.13 mmol / l (normal range: < 1.7 mmol / l), fibrinogen 1.43 g / l (normal range: 2-4 g / l). Ferritin 555.21 ng / ml (normal range: 21.8 - 274.6 ng / ml). Blood TORCH virus antibody: herpes simplex virus antibody IgG (+), rubella virus antibody IgG (+), cytomegalovirus antibody IgG (+). EB virus antibody IgG (+), EB-DNA copies were normal. Tumour markers: carcino embryonic antigen (CEA) 6.61 ng / ml (normal range: < 5 ng / ml), carbohydrate antigen 125 (CA125): 93.27 u / ml (normal range: < 35 u / ml.
Cerebro Spinal Fluid (CSF): WBC 5 × 10^6 / l (normal range: 0 - 8 × 10^6 / l), protein 0.58 g / l (normal range: 0.15 - 0.45 g / l), sugar 3.24 mmol / l (normal range: 2.3 - 4.1 mmol / l), chlorine 127 mmol / l (normal range: 119 - 129 mmol / l), Pan’s reaction (+). Cytology: A small amount of neutrophils and lymphocytes and no abnormal cells. Blood soluble interleukin-2 receptor (sil-2r): 786 u / ml (normal range: 223 - 710 u / ml), NK cell (CD3-CD56+): 2% (normal range: 8.75 - 40.52%), CMV-PP65 (+). ESR and blood culture showed no abnormalities. Human Immunodeficiency Virus (HIV) antibody was negative, Rapid Plasma Regain (RPR) was nonreactive, and hepatitis B and C virus serologies were negative. Anti-Nuclear Antibody (ANA) and anti-double-stranded DNA were negative, rheumatoid factor was negative, complement levels were normal, and both cytoplasmic and perinuclear anti-neutrophil cytoplasmic antibodies (c-ANCA and p-ANCA) were negative. TB-Ab and the Brucella agglutination test were negative. Cancer cells, Cryptococcus, or Mycobacterium tuberculosis were not found in the CSF samples. Autoimmune encephalitis antibody and paraneoplastic syndrome antibody were negative. Bone marrow biopsy indicated granulocyte maturation disorder, an increase in plasma particles, megakaryocyte maturation disorder, and haemophagocytosis (Figure 4). A heterozygous mutation point was found in the exon region of the patient’s FTL gene: c.123delt (deletion mutation), resulting in amino acid changes p.d41fs (frame shift mutation).
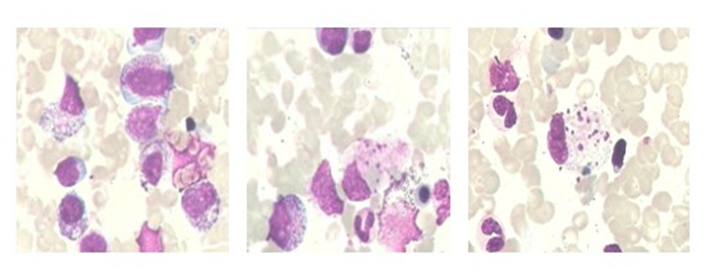
Figure 4: Bone marrow smear (2016.05.12): Granulocyte maturation disorder, plasma particles increase; Megakaryocyte maturation disorder; Bloodthirsty phenomenon can be seen. (as arrow indicated )
The patient was finally diagnosed with CNS-HPS. As the patient’s CSF cell population was normal and his CSF antibody tests were negative, encephalitis and meningitis cannot be diagnosed. The patient was treated with intravenous immunoglobulin (0.4 g / kg / day for 5 days), dexamethasone (10 mg / day intravenous for 14 days), anti-inflammatory medications (cefminox, 0.2 g / q8h for 15 days), antiviral medications (acyclovir, 0.5 g / q8h for over 14 days), and symptomatic and supportive care. After 15 days of treatment, the patient’s symptoms were relieved. His body temperature returned to normal and his seizures ceased, but he remained unconscious with marked hypermyotonia. After one month, the patient regained consciousness and basic communication skills and his muscle tension improved. A routine blood test and liver function test were normal. A bone marrow biopsy showed no haemophagocytosis (Figure 5). A brain MRI showed the range of lesions had significantly reduced, and some lesions had disappeared (Figure 6). After two months, the patient had only distal weakness of the lower limbs and there were no abnormalities in his laboratory examination results. He was transferred from the neurology department to the rehabilitation department for further treatment.
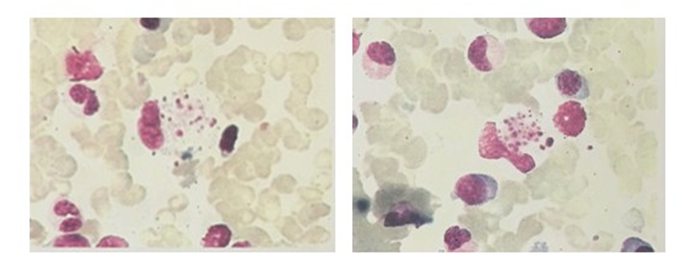
Figure 5: Bone marrow smear (2016.06.16): Bone marrow hyperplasia, granulocyte granules increased significantly, phagocytic cells can be seen, considering the infection.
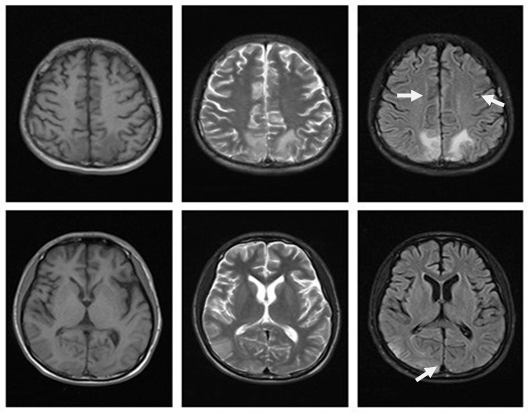
Figure 6: brain MRI(2016-06-28): The bilateral parietal lobe, right temporal lobe and occipital lobe show patch equal T1 signal and slightly long T2 signal, flair phase shows hyperintense. Compared with the primary images, the range of lesions was significantly reduced, some lesions disappeared(as the arrows indicated).
Discussion
Hemo Phagocytic Syndrome (HPS) is a rare disorder that is characterized by proliferation of benign histiocytes, and this commonly involves the liver, spleen, lymph nodes, bone marrow and central nervous system (CNS)[9]. The frequency of CNS involvement in HLH is unclear, especially in adults. In one small series CNS involvement was identified in up to 73% of patients[10]. In another, CNS involvement was present in 30% of patients who had both HLH and an underlying autoimmune disorder[11]. When HPS is involved in the central nervous system, acute organic brain syndrome can be observed in the early stage of the disease, which is characterized by dizziness, headache, sleep disorders and other neurasthenia symptoms. Late stage, such as memory disorders, emotional apathy, lethargy, cranial nerve palsy, seizures, ataxia, disturbance of consciousness, elevated intracranial pressure and other neuropsychiatric symptoms[12]. The CT findings include diffuse parenchymal atrophy, low attenuated lesions in the white matter, and calcifications[13,14]. The MR findings include diffuse leptomeningeal and perivascular enhancement and a diffuse parenchymal volume loss of the cerebrum and cerebellum. In some cases, nodular or ring enhancement of the parenchymal lesion appears due to the compromised blood-brain barrier that is associated with active demyelination[15]. The pathology of HPS nervous system damage is mainly the infiltration of lymphocytes and macrophages in the meninges, blood vessels, peripheral tissues and brain tissues, and tissue diffusion and necrosis caused by south infiltration.
According to the diagnostic guidelines for HLH-2004[17], HPS is clinically diagnosed by either:
Having a proven genetic mutation known to be associated with HPS or
Fulfilling 5 out of the following 8 clinical criteria:
• Fever
• Splenomegaly
• Cytopenia affecting at least two of three lineages
• Hypertriglyceridemia and/or hypofibrinogenaemia
• Haemophagocytosis
• Low/absent NK-cell activity
• Hyperferritinaemia and
• High-soluble interleukin-2-receptor (soluble CD25) levels[7].
The patient had 5 of the 8 clinical criteria: splenomegaly, pancytopenia, hypertriglyceridemia, and haemophagocytosis. Consistent with the clinical manifestations of HPS. Brain MRI showed the bilateral frontal lobe, occipital lobe, parietal cortex and subcortical white matter multiple abnormal signal. CSF protein was slightly increased. These proved the involvement of the central nervous system. Thus it was finally diagnosed as CNS-HPS. The neuro imaging presentation of HPS is not specific. The MRI features may mimic those of neoplastic, infectious, or inflammatory disorders[14], so it is easy to misdiagnose. In the present case, the patient’s brain MRI identified Reversible Posterior Leukoencephalopathy Syndrome (RPLS), which demonstrates the most prominent lesions distributed in the posterior cerebral circulation area, bilateral occipital cortex, and subcortical white matter. Most imaging changes are reversible, and lesions can disappear after 2 weeks or longer. Bone and peripheral blood tests often help the diagnosis of HLH.
There is a mutation in the FTL gene in his and his patient’s genetic test. His mother has a mutation site. Considering the possibility of FHLH. FHL can be divided into 5 types: FHLH1-5, immunodeficiency syndrome-related HLH includes three syndromes: Chediak Higashi syndrome (CHS), Griscelli Syndrome 2 (GS2) and X-linked lymph proliferative disease (XLP). Primary HPS usually manifests as a result of genes defects encoding the proteins involved in the exocytosis of cytotoxic granules in Natural Killer (NK) cells, which leads to hyper activation of the immune system[3]. Genes encoding for perforin, syntaxin 11, Munc 13-4, and Munc 18 - 2 proteins have been implicated[18]. FHLH has widely been considered as a disease characteristic of the first two years of age, Since the original report of two affected siblings aged nine weeks by James Farquhar in 1952, which may mislead the clinician to be afraid of diagnosing FHLH in adults with more and more sporadic reports of FHLH in older children and adolescents, a possible ‘threshold’ age for diagnosing FHLH have raised the issue[19].
HPS is rare in adults and CNS involvement is even rarer, so adult-onset CNS-HPS is easily missed and misdiagnosed. Therefore, imaging findings must be correlated with suspicious clinical and laboratory abnormalities such as fever, hepatosplenomegaly, neurologic symptoms, rash, pancytopenia, hypertriglyceridemia, hyperferritinaemia, and hypofibrinogenaemia. This should be considered in the differential diagnosis of adults presenting with progressive brain lesions, even in the absence of typical systemic signs of HPS. Bone marrow biopsy is the key to an early diagnosis. Clinicians should improve their understanding of the disease, early diagnosis, and timely treatment as these are vitally important for a satisfactory prognosis and reduction in the mortality of Haemophagocytic Syndrome.
Conflict of Interest: The authors contributed equally to this work and declare that they have no competing financial interests.
References
- 1. Janka, G.E., Lehmberg, K. Hemophagocytic lymphohistiocytosis: pathogenesis and treatment. (2013) Hem Am Soc Hem Edu Pro: 605-11.
- 2. Mehta, R.S., Smith, R.E. Hemophagocytic lymphohistiocytosis (HLH): a review of literature. (2013) Med Onco 30(4): 740.
- 3. Rosado, F.G., Kim, A.S. Hemophagocytic lymphohistiocytosis: an update on diagnosis and pathogenesis. (2013) Am J Clin Pathol 139(6): 713-727.
- 4. Chandrakasan, S., Filipovich, A.H. Hemophagocytic lymphohistiocytosis: advances in pathophysiology, diagnosis, and treatment. (2013) J Pediatr 163(5): 1253-1259.
- 5. Gurgey, A., Aytac, S., Balta, G., et al. Central nervous system involvement in Turkish children with primary hemophagocytic lymphohistiocytosis. (2008) J Child Neurol 23(11): 1293-1299.
- 6. Deiva, K., Mahlaoui, N., Beaudonnet, F., et al. CNS involvement at the onset of primary hemophagocytic lymphohistiocytosis. (2012) Neurology 78(15): 1150-1156.
- 7. Piccin, A., Raimondi, R., Laspina, S., et al. Erythroleukaemia, diabetes insipidus and hypophyseal damage: Two case reports. (2007) Leuk Res 31(8): 1135-1139.
- 8. Sieni, E., Cetica, V., Piccin, A., et al. Familial hemophagocytic lymphohistiocytosis may present during adulthood: clinical and genetic features of a small series. (2012) PLoS One 7(9): 44649.
- 9. Henter, J.I., Elinder, G., Söder, O., et al. Incidence in Sweden and clinical features of familial hemophagocytic lymphohistiocytosis. (1991) Acta Paediatr Scand 80(4): 428-435.
Pubmed || Crossref || Others
- 10. Janka, G.E. Familial and acquired hemophagocytic lymphohistiocytosis. (2007) Eur J Pediatr 166(2): 95-109.
- 11. Haddad, E., Sulis, M.L., Jabado, N., et al. Frequency and severity of central nervous system lesions in hemophagocytic lymphohistiocytosis. (1997) Blood 89(3): 794-800.
- 12. Fukaya, S., Yasuda, S., Hashimoto, T., et al. Clinical features of haemophagocytic syndrome in patients with systemic autoimmune diseases: analysis of 30 cases. (2008) Rheumatology (Oxford) 47(11): 1686-1691.
- 13. Anderson, T.L., Carr, C.M., Kaufmann, T.J. Central nervous system imaging findings of hemophagocytic syndrome. (2015) Clin Imaging 39(6): 1090-1094.
- 14. Munoz, R.M.M., Castillo, M. Brain CT and MR imaging in familial hemophagocytic lymphohistiocytosis. (1998) AJR Am J Roentgenol. 170(3): 802.
Pubmed || Crossref || Others
- 15. Forbes, K.P., Collie, D.A., Parker, A. CNS involvement of virus-associated hemophagocytic syndrome: MR imaging appearance. (2000) AJNR Am J Neuroradiol 21(7): 1248-1250.
Pubmed || Crossref || Others
- 16. Henter, J.I., Nennesmo, I. Neuropathologic findings and neurologic symptoms in twenty-three children with hemophagocytic lymphohistiocytosis. (1997) J Pediatr 130(3): 358-365.
- 17. Henter, J.I., Horne, A., Aricó, M., et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. (2007) Pediatr Blood Cancer 48(2): 124-131.
- 18. Cetica, V., Pende, D., Griffiths, GM., et al. Molecular basis of familial hemophagocytic lymphohistiocytosis. (2010) Haematologica 95(4): 538-541.
- 19. Trottestam, H., Horne, A., Aricò, M., et al. Chemo immunotherapy for hemophagocytic lymphohistiocytosis: long-term results of the HLH-94 treatment protocol. (2011) Blood 118(17): 4577- 4584.










