
Cellular Responses in Titanium Dioxide Nanoparticle Cytotoxicity Studies: Parts of the Map Waiting to be Composed
Shunbang Yu1, Joanna Bohatko-Naismith1, Xudong Zhang2, Xiaonan Zhou3, Pu Wang4, He Wang1*
Affiliation
- 1School of Health Sciences, University of Newcastle, Callaghan, NSW 2308, AUSTRALIA
- 2School of Biomedical Sciences and Pharmacy, University of Newcastle, Callaghan, NSW 2308, AUSTRALIA
- 3School of Nursing, Peking Union Medical College, Beijing 100144, CHINA
- 4School of Clinical, Baotou Medical College, Baotou, Inner Mongolia Autonomous Region 014040, CHINA
Corresponding Author
Yu, S., School of Health Sciences, University of Newcastle, Callaghan, NSW 2287, Australia, E-mail: c3220962@uon.edu.au
Citation
He Wang., et al. Cellular Responses in Titanium Dioxide Nanoparticle Cytotoxicity Studies: Parts of the Map Waiting to be Composed. (2017) J Med Chem Toxicol 2(2): 62- 70.
Copy rights
© 2017 Yu, S. This is an Open access article distributed under the terms of Creative Commons Attribution 4.0 International License.
Keywords
Titanium Dioxide Nanoparticle; Cytotoxicity; Apoptosis; Senescence; p53; Inflammation
Abstract
With the infiltration of nanotechnology into our daily life, exposure to Titanium Dioxide Nanoparticles (TNPs) has become inevitable. Many forms of exposures are capable of affecting the human health. Current research in the Titanium dioxide nanoparticle pathology field reveals that TNP is capable of inducing severe oxidative stress to in vivo and in vitro cells, which predominantly result in cellular apoptosis. This article summarizes the main pathways of TNP induced apoptosis and compares it with cigarette-smoke-induced senescence, in an attempt to identify the similarities and acknowledge knowledge gaps to question the integrity of the TNP cytotoxicity studies conducted. The viable cells in the studies reviewed are not defined, and may provide a direction for future TNP toxicology studies.
Introduction
TNPs (φ < 100 nm) are widely used in photocatalyst cleaning of water and air[37], cosmetics, toothpaste, sunscreen, and pharmaceuticals[38]. While TiO2 itself may have proved to hold a very broad future in material science and relevant industries, nano-sized TiO2 particles have been proved to induce greater toxicity than larger particles due to its more active chemical properties, greater surface area and easier cell membrane penetration[39,40] and extensive studies have proved that TNPs are capable of inducing apoptosis in various types of cell cultures and live animal tissue cells. The molecular mechanism and processes of the cellular apoptosis induced by TNPs remain under-studied. However, multiple pathways have been observed and specified. These regulation pathways include:
1. Upregulation of caspase -3 and -9 [2,4,5,26-32.,41-44] and p53[2-5,29] activation, upregulation of the BAX/Bcl-2 ratio[2,4,5,21,27-31,33,44-47] and inflammatory kinases[26,34,35,48,49] in the mitochondrial (intrinsic) pathway.
2. Caspase-8 production, Bid activation[27,34] and death receptor Fasactivation[28,50-52] in the death receptor (extrinsic) pathway.
This article summarizes the defined pathways of TNP induced apoptosis and compares it with the senescence process of cells (cigarette smoke as the stress factor due to its universality), hence providing guidance for future research and contribute to a more comprehensive understanding of TNP cytotoxicity regarding characterization of the overall response of the cell economy to TNP cytotoxicity.
Intrinsic (mitochondrial) pathway
The impairment of energy metabolism has been extensively implicated in cellular pathologies. Dysfunctional mitochondria is a major factory of ROS production. In the studies of TNP pathologies, researchers have identified the impairment of mitochondria as a major factor of apoptosis. However, the specific mechanism of ROS production resulting from TNP stress remains understudied and awaits further investigations.
1. DNA damage and p53 upregulation
The involvement of tumour suppressor p53 in TNP induced apoptosis has been extensively studied in recent years. DNA damage resulting from TNP induced oxidative stress leads to the activation of p53[2-5,29,44,53,54], which triggers cell cycle arrest to prevent proliferation of cells with damaged DNA and ensure genomic stability[55]. In response to apoptosis inducing stress, p53 rapidly translocates to the mitochondria, where it interacts with the Bcl-2 family members, consequently upregulating the pro-apoptotic proteins which include Mitochondrial Outer Membrane Permeability (MOMP) effectors BAX and Bak (formation of lipid pores on the mitochondria outer membrane), apoptosis activators tBid and Bim (stimulation of BAX and Bak oligomerization), and anti-apoptotic protein inhibitor Puma, Noxa, and Bad (inhibition of anti-apoptotic Bcl-2 family members)[55,56].
2. ROS production
The production of ROS has been regarded as the initiator of the mitochondria pathway of TNP induced apoptosis. Many researchers have made observations of the pro-apoptotic process. It is speculated that the TNPs induce the production of ROS through disrupting the natural redox (reduction-oxidation) equilibrium[28]. The specific mechanisms of this process remains unknown, however, speculations could be made in reference to studies based on the cytotoxicity of other nanomaterials. Kee Woei Ng., et al. proposed in their study on Zinc Oxide (ZnO) Nanoparticle (ZNP) cytotoxicity that ZnO produces Zn2+ upon intracellular and extracellular dissolution[10]. The Zn2+s induce the production of ROS through an unknown mechanism. Molecular analysis has identified complex I and III as the main generators of free radicals in the electron transport chain in hyperactive mitochondria[57]. Researchers have confirmed that the oxidative stress induced by metallic anions results in ROS production, but the specific mechanism awaits further definition. With the capability of snatching electrons away from biomolecules[57], ROS are capable of inflicting damages such as cell and mitochondria membrane lipid peroxidation[4,5,29,45,58-67] and increased permeability of the outer mitochondrial membrane[4,27,68-72]. Yongli Shi., et al. demonstrated a highly relevant relationship between the amount of ROS production, apoptosis intensity, and cell viability in BEAS-2B cell lines (human bronchial epithelium cell culture) treated with TNPs (Figure 1). Statistical analysis indicates that TNP exposure intensity, ROS production, apoptosis rate (Napoptotic % ≈ 1 ˗ Nviable %) are engaged in a relationship definable with function (Figure 2). However, the state of the cells classified to possess viability wasn’t described. Similar findings can be found in the work of Jinshun Zhao., et al where TNP exposure was introduced to JB8 cell culture[27]. As it is presented in the figures (figures 3,4 5), while a large proportion of BEAS-2B cells exhibited apoptosis (reaching 30 % with the increase of TNP exposure intensity), a greater number of cells maintained viability, which contradicts the theory that epithelial cells respond to stress primarily via apoptosis[20,36]. Furthermore, it is unclarified what type of viable state the viable cells exhibit and the description of the whole cell economy may be of great importance in explaining and defining TNP toxicity.
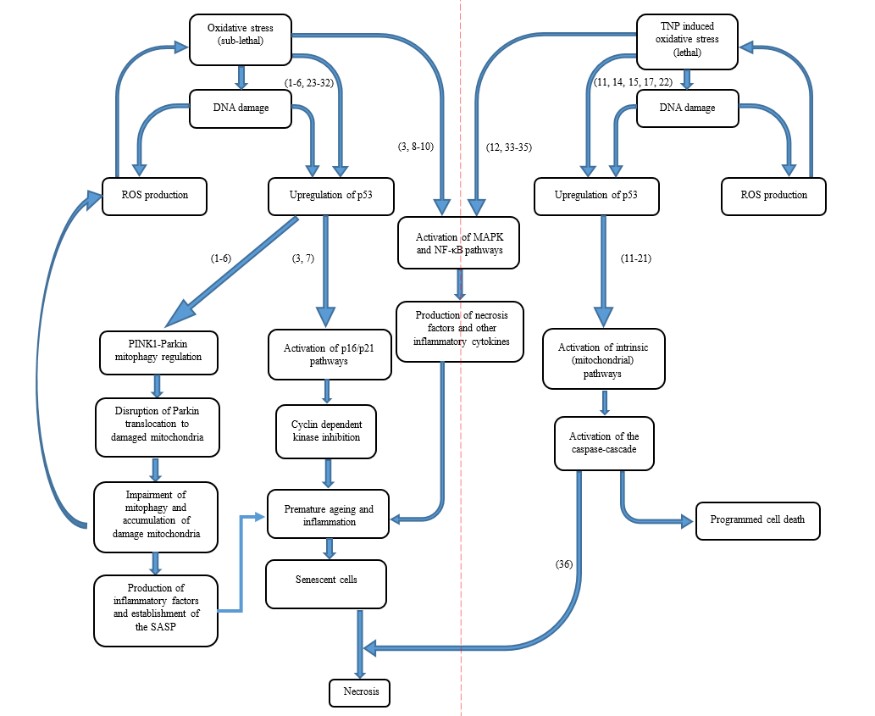
Figure 1: Overall process and relationship between different cellular pathologies relevant to this review.
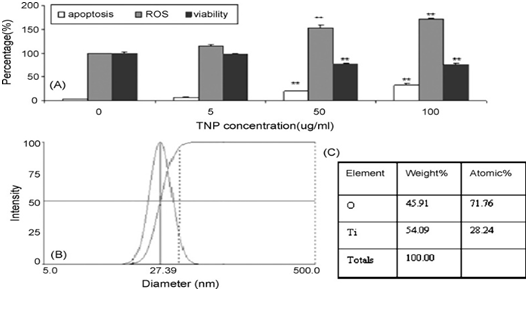
Figure 2: Yongli Shi et al. presented this figure in their papers to clarify their observations of TNP exposure affecting BEAS-2B cellular responses(2). (A) Measurement of apoptotic cells (YP staining), ROS generation, and viable cells (MTT assay) in cells treated with different concentrations of TNP for 24 hrs. (B) DLS assay for TNP. (C) Element analysis of TNP.
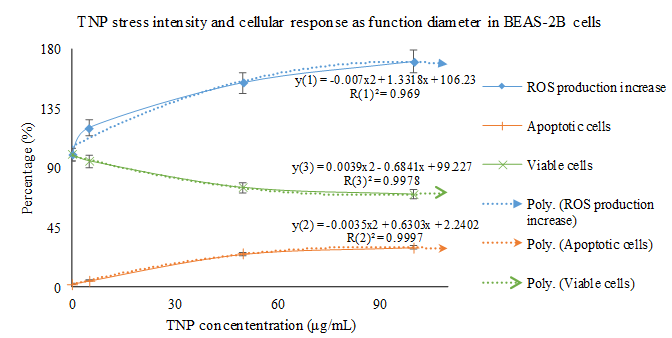
Figure 3: Statistical analysis reveals that the cellular responses (in BEAS-2B cells) to TNP stress is in a linear relationship. It is noticeable that cell death occurs upon the administration of TNP stress, even at a very low intensity. However, the cells possessing viability have not been characterized. The trend line of ROS production, apoptotic cell count, and viable cell count related with TNP stress intensity are referred to as y(1), y(2), and y(3), respectively.
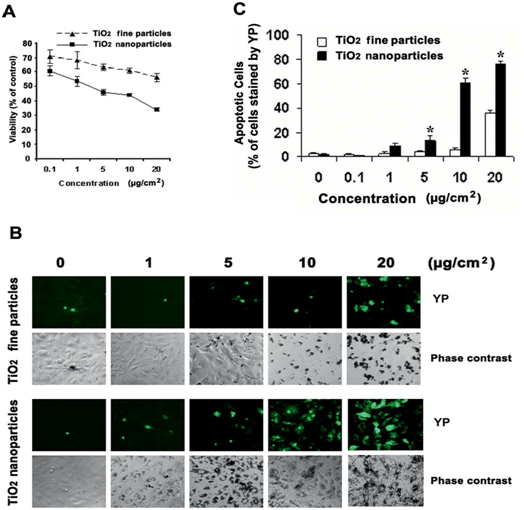
Figure 4: Similar to the results presented by Yongli Shi et al., Jinshun Zhaoet al. demonstrated the same trend in JB8 cells. However, the JB8 cells were more sensitive to TNP stress, and have been observed with a higher occurrence rate of apoptosis compared to that of the BEAS-2B cell lines under a lower intensity of TNP exposure. Additionally, the occurrence rate of cellular response was more abrupt and un-foreseeable compared to that exhibited by the BEAS-2B cells (Figure. 4). (A) MTT assay was conducted.
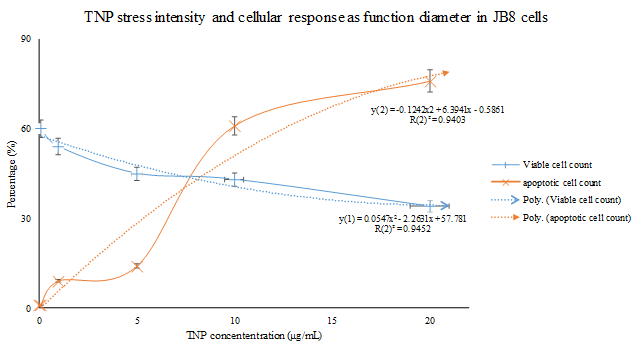
Figure 5: Statistical analysis reveals that the sensitiveness of JB8 cells to changes in TNP exposure intensity fluctuates across a relatively smaller range (compared with the analysis done in Figure.2). The cause of this difference wasn’t defined.
3. Lipid peroxidation and increased membrane permeability
A major damage induced by TNP induced ROS production is lipid peroxidation of the cellular and mitochondrial membranes. Lipid peroxidation of mitochondrial membrane leads to dysfunctions of the mitochondria, and uncontrolled opening of the outer membrane transition pores have been considered as an important pro-apoptotic response. Uncontrolled opening of the transition pores is a major cause for increased permeability of the mitochondria, translocation of cytosolic apoptotic proteins into the mitochondria, and leakage of mitochondrial contents into the cytosol[54]. Researchers have observed the increase of cytochrome c in the cytosol[2,27,43,45,50,71,73,74]. This was mainly a result of peroxidation of membrane lipid and phospholipid Cardiolipin (CL), which binds with cytochrome c and anchors it to the inner mitochondrial membrane to participate in electron transport[57,73]. In earlier studies, Paradies G., et al. demonstrated that mitochondrial ROS induces a CL decrease, leading to decrease of cytochrome c oxidase, which is reliant on the presence of CL. This was repairable with added CL, but not with addition of peroxidised CL nor antioxidant enzymes such as Superoxide Dismutase (SOD) and catalase[75]. Once translocated into the cytosol, cytochrome c is capable of binding to the critical protein Apaf-1, which then binds to and activates caspase-9[54], consequently resulting in activation of caspase-3 and apoptosis induction[2,27,28].
Extrinsic (death receptor) pathway
1. The Fas/caspase-8 pathway
The Fas-Fas ligand (FasL) signalling is an important member of the extrinsic apoptosis pathways. In the normal cell, Fas-FasL signalling pathway acts to initiate apoptosis when FasL is produced by T cells and activates the Fas death receptor on target cells, with leads to the activation of the caspase-3 and caspase-8[24]. Fas death receptor activation was observed by Ki-Chun Yoo., et al. in different kinds of human cell lines treated with TNPs[28]. However, Fas ligand production was not mentioned, and ROS was implicated in the activation of Fas death receptors instead. However, no description to how ROS mediates Fas activation has been given, and further research awaits initiation.
2. Tumour necrosis factor-α induced cell death
The TNF-α is a cytokine that is associated with severe inflammation and triggers necrosis-like apoptosis mainly in endothelium and lymphoid tissues[54]. Apart from being able to trigger caspase-8 activation, it is also capable of binding with its specific death receptor and initiating caspase-independent apoptosis. Researchers have confirmed the role of TNP in TNF-α induction in various kinds of human cells[26,35,47,76,77]. This may provide an explanation that although TNP was proved to predominantly induce apoptosis, inflammation and even emphysema-like pathologies was also observed in TNP treated cells[26,31,34,37,47,71,78,79].
An attempt to adapt and survive – inflammatory responses
During research on TNP cytotoxicity, Eun-Jung Park., et al. observed the upregulation of a series of inflammatory factors including interleukin- (IL-) 1, 6, 8, and TNF-a and CXC motif ligand 2 (CXCL-2) in TNP treated human bronchial epithelial cells[26]. During their experiments, IL-8 upregulation was prevented with the inhibition of the p-38 Mitogen-Activated Protein Kinase (p38-MAPK) and Extracellular Signal-Regulated Kinase (ERK) pathway, which provides evidence for the activation of these two inflammatory pathways in the TNP induced apoptosis process. What should be noticed is that MAPK and ERK, along with the c-Jun NH2-terminal kinase (JKN1) and Apoptosis Signal-Regulating Kinase 1 (ASK1) are inflammatory kinases that are implicated in anti-apoptotic mechanisms[54]. In addition, Jin Chen., et al. also observed NF-κB upregulation in TNP treated hepatocytes[34]. Among the inflammatory factors mentioned, although the MAPK and NF-κB pathways have also been implicated in cell survival or cellular senescence, no detections of cellular senescence have been made as a cellular response to TNP cytotoxicity. It could be observed that TNP treated cells share some similar characteristics with senescent cells, and are proved to bepro-inflammatory in the studies. Whether this can lead to carcinogenic responses awaits further investigation.
A brief review on stress induced premature senescence
Senescence is a state of the cell characterized by entering an irreversible cell cycle arrest. Cells under the senescent state exhibit a high secretory activity of inflammatory chemokines known as the senescence associated secretory phenotype (SASP)[80]. Although multiple pathways of senescence initiation have been observed, the specific explanation for how senescence is molecularly mediated still remains unknown. What should be noticed is that the molecular responses that have been observed be induced by TNPs are also associated with cellular senescence establishment.
The p53 related pathway
Tumour suppressor p53 has been reported to be greatly associated with senescence establishment in cigarette smoke exposure experiments. The upregulation of p53 was reported by researchers as the result of DNA damage[6,8,9,11,12,14-1620,21]. However, the downstream factors of the p53 pathways is completely different in comparison with the p53 related pathways in apoptosis induction.
1. The p53-Parkin interaction
In the normal autophagic activities of the cell, the PINK1-Parkin (or PARK2) pathway is implicated in the clearance of damaged mitochondria. PINK1 immobilizes to mitochondria with depolarized membrane, and induces the recruitment of Parkin to the damaged mitochondria’s outer membrane[14,16]. There, Parkin initiates the degradation of mitofusin-2 (Mfn2, a protein implicated in the fusion of mitochondria), therefore preventing mitochondrial fusion and initiating the formation of autophagosomes, which triggers mitophagy. During exposure to cigarette smoke, Tanveer Ahmad., et al. observed that the targeted autophagy of damaged mitochondria (mitophagy) was greatly impaired due to the interaction between p53 and Parkin, thus inhibiting the translocation of Parkin to damaged mitochondria. This was supported by his observations of perinuclear accumulation of damaged mitochondria after cigarette smoke treatment, which ultimately resulted in enhancement of senescence[14]. This may be one of the sources of the irreversibility and progressiveness of pulmonary-inflammation-related and premature-ageing-related diseases.
2. The ATM/ATR-p53-p21-pRb pathway
In the majority of somatic cells, DNA Damage Response (DDR) activates the sensor kinases ATM/ATR, which results in phosphorylation of multiple downstream players of the signalling cascade, including H2AX histone at the site of DNA breaks, and transcription factor p53. The former results in formation of γ-H2AX foci, which is implicated in the recruitment and anchoring of DDR players. The latter results in transcription of various genes including p21 and GADD45, which are capable of inhibiting cyclin-dependent kinases. Then, resulting from effects of various stressors, initiation of the p38/MAPK cascade activates the p16-pRB (retinoblastoma protein). As inhibitors of cyclin dependent kinase, p21 and p16 mediates the hypophosphorylation of RB, which binds to transcription factor E2F and affects cell cycle progression[15,81,82]. Similarly, Yi Luo., et al. observed that autophagy is capable of inducing senescence via p21 through a p38-MAPKα dependent pathway. The above observations contribute to the explanations of the inflammation observed in TNP treated cells, and provides support to speculate that senescence is a great possibility of an attempt at cell survival in TNP treated cells given the conditions are right.
Inflammatory induction and carcinogenic effects of premature ageing
Senescent cells possess a degree of metabolic and transcriptional vitality, therefore capable of affecting the micro-environment. Furthermore, although cellular senescence have been originally regarded as an anti-tumour and cytoprotective response of the cell, recent research reveals that chronic premature senescence in the cell economy and tissues may be carcinogenic through secretion of the SASP[80]. Jean-Philippe Coppé., et al. concluded in their research that the SASP could be divided into four categories, including soluble factors, soluble or shed receptors or ligands, non-protein soluble factors, and insoluble factors. Among those regulations, the upregulation of certain interleukins (e.g. IL-1, -6, -8), inflammatory cytokines and chemokines (e.g. PlGF, CXCL1, CXCL5, CCL2/MCP-1, CCL3/MIP-1) and ROS were similar to that of the regulation pattern of TNP induced poptosis[37]. Apoptosis in addition, exhibited a high production of apoptotic factors such as caspases[27].
In terms of inflammation, the MAPK and NF-κB pathways are also a great contributor in the inflammatory effects of premature ageing.
Although researchers claim that senescence and apoptosis may be different responses of the cells reacting differently to different levels of stress exposed to different cell types[20,36], they have also proposed that senescence is a cytoprotective response of the cell initiated in chronic stress to maintain cell economy and prevent over-loss and damage of the tissue from apoptosis. Eleni Georgakopoulou., et al. proposed a model for carcinogenesis, in which stroma cells are not directly affected by the stressors acting on the epithelium, but rather identifies cancer as a wound that does not heal. While fibroblasts transiently proliferate to maintain structural stability and cell economy, persistent lesions cause stroma fibroblasts to become senescent in order to prevent fibrosis. However, no research have clearly proved whether this model suits the TNP pathologies.
Knowledge Gap and Discussion
It is reasonable to conclude that the cellular response described in previous research on TNP toxicology was predominantly apoptosis. However, the changing rate of the apoptosis proportion trended towards zero after a certain intensity of stress, potentially meaning a proportion of the counted cells are capable of maintaining viability. We know that cells undergone stress can exhibit different kinds of “being viable”, such as cell survival, dormancy and senescence. How the viable cells responded to TNP cytotoxicity wasn’t analysed, and the difference of possibilities may greatly affect the systematic definition of TNP toxicology, providing guidance and reference for future nanoparticle toxicology studies and pharmaceutical applications of nanoparticles.
Secondly, current research on TNP toxicology lacks comparison of key intracellular senescence and apoptosis biomolecules. Specifically, current research in the field doesn’t discuss the potential interactions between different cellular responses in the same tissue economy, but rather individually discuss one as the predominant cellular response resulting from a specific type and intensity of stress. Observations of pre-apoptotic intracellular molecule regulations in senescence induction and pre-senescence intracellular molecule regulations in apoptosis induction may provide a broader view in understanding how the cell decides the way out. For example, some researchers reported that upregulation of induced autophagic activity in senescent cells resulted in the induction of apoptosis, while downregulation of induced autophagy and upregulation of baseline autophagy contributed to the establishment of cellular senescence and SASP secretion[8,16,83]. Contrarily, Hongmei Dai., et al. proposed that the PINK1-Parkin-mediated mitophagy alleviates apoptosis in human SH-SY5Y cell lines treated with chlorpyrifos[84].
In conclusion, although extensive work has been conducted to study TNP cytotoxicity, further investigation that is based on a comprehensive mapping of the cell economy and the associated cellular responses is necessary.
Conflict of interest:
The authors declare no conflict of interest.
References
- 1. Wang, J., Zhou, G., Chen, C., et al. Acute toxicity and biodistribution of different sized titanium dioxide particles in mice after oral administration. (2007) Toxicol Lett 168(2): 176-185.
Pubmed || Crossref || Others - 2. Shi, Y., Wang, F., He, J., et al. Titanium dioxide nanoparticles cause apoptosis in BEAS-2B cells through the caspase 8/t-Bid-independent mitochondrial pathway. (2010) Toxicol Lett 196(1): 21-27.
Pubmed || Crossref || Others - 3. Wu, J., Sun, J., Xue, Y. Involvement of JNK and P53 activation in G2/M cell cycle arrest and apoptosis induced by titanium dioxide nanoparticles in neuron cells. (2010) Toxicol Lett 199(3): 269-276.
Pubmed || Crossref || Others - 4. 4. Meena, R., Rani, M., Pal, R., et al. Nano-TiO2-induced apoptosis by oxidative stress-mediated DNA damage and activation of p53 in human embryonic kidney cells. (2012) Appl Biochem Biotechnol 167(4): 791-808.
Pubmed || Crossref || Others - 5. Shukla, R.K., Kumar, A., Gurbani, D., et al. TiO(2) nanoparticles induce oxidative DNA damage and apoptosis in human liver cells. (2013) Nanotoxicol 7(1): 48-60.
Pubmed || Crossref || Others - 6. Tasdemir, E., Maiuri, M.C., Galluzzi, L., et al. Regulation of autophagy by cytoplasmic p53. (2008) Nat Cell Biol 10(6): 676-687
Pubmed || Crossref || Others - 7. Salminen, A., Kaarniranta, K. Regulation of the aging process by autophagy. (2009) Trends Mol Med 15(5): 217-224.
Pubmed || Crossref || Others - 8. Young, A.R., Narita, M., Ferreira, M., et al. Autophagy mediates the mitotic senescence transition. (2009) Genes Dev 23(7): 798-803.
Pubmed || Crossref || Others - 9. Luo, Y., Zou, P., Zou, J., et al. Autophagy regulates ROS-induced cellular senescence via p21 in a p38 MAPKα dependent manner. (2011) Exp Gerontol 46(11): 860-867.
Pubmed || Crossref || Others - 10. Ng, K.W., Khoo, S.P., Heng, B.C., et al. The role of the tumor suppressor p53 pathway in the cellular DNA damage response to zinc oxide nanoparticles. (2011) Biomaterials 32(32): 8218-8225.
Pubmed || Crossref || Others - 11. Fujii, S., Hara, H., Araya, J., et al. Insufficient autophagy promotes bronchial epithelial cell senescence in chronic obstructive pulmonary disease. (2012) Oncoimmunology 1(5): 630-641.
Pubmed || Crossref || Others - 12. Yao, H., Chung, S., Hwang, J.W., et al. SIRT1 protects against emphysema via FOXO3-mediated reduction of premature senescence in mice. (2012) J Clin Invest 122(6): 2032-2045.
Pubmed || Crossref || Others - 13. Goehe, R.W., Bristol, M.L., Wilson, E.N., et al. Autophagy, Senescence, and Apoptosis. (2013) Methods Mol Biol 962: 31-48.
Pubmed || Crossref || Others - 14. Ahmad, T., Sundar, I.K., Lerner, C.A., et al. Impaired mitophagy leads to cigarette smoke stress-induced cellular senescence: implications for chronic obstructive pulmonary disease. (2015) FASEB J 29(7): 2912-2929.
Pubmed || Crossref || Others - 15. Fridlyanskaya, I., Alekseenko, L., Nikolsky, N. Senescence as a general cellular response to stress: A mini-review. (2015) Exp Gerontol 72: 124-128.
Pubmed || Crossref || Others - 16. Ito, S., Araya, J., Kurita, Y., et al. PARK2-mediated mitophagy is involved in regulation of HBEC senescence in COPD pathogenesis. (2015) Autophagy 11(3): 547-559.
Pubmed || Crossref || Others - 17. Lasry, A., Ben-Neriah, Y. Senescence-associated inflammatory responses: aging and cancer perspectives. (2015) Trends Immunol 36(4): 217-228.
Pubmed || Crossref || Others - 18. Mytych, J., Pacyk, K., Pepek, M., et al. Nanoparticle-mediated decrease of lamin B1 pools promotes a TRF protein-based adaptive response in cultured cells. (2015) Biomaterials 53: 107-116.
Pubmed || Crossref || Others - 19. Nadezhda, V. Petrova1 AKV, Natalia V. Petrova1, Sergey V. Razin1,2, Omar L. Kantidze1. Stress factor - dependent differences in molecular mechanisms of premature cell senescence. (2015) Biopolymers and Cell 31: 323-337.
Pubmed || Crossref || Others - 20. Georgakopoulou, E., Evangelou, K., Havaki, S., et al. Apoptosis or senescence? Which exit route do epithelial cells and fibroblasts preferentially follow? (2016) Mech Ageing Dev 156: 17-24.
Pubmed || Crossref || Others - 21. McCubrey, J.A., Lertpiriyapong, K., Fitzgerald, T.L., et al. Roles of TP53 in determining therapeutic sensitivity, growth, cellular senescence, invasion and metastasis. (2017) Adv Biol Regul 63: 32-48.
Pubmed || Crossref || Others - 22. Baker, D.J., Wijshake, T., Tchkonia, T., et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. (2011) Nature 479(7372): 232-236.
Pubmed || Crossref || Others - 23. Tsuji, T., Aoshiba, K., Nagai, A. Alveolar cell senescence exacerbates pulmonary inflammation in patients with chronic obstructive pulmonary disease. (2010) Respiration 80(1): 59-70.
Pubmed || Crossref || Others - 24. Li, Z., Li, J., Bu, X., et al. Age-induced augmentation of p38 MAPK phosphorylation in mouse lung. (2011) Exp Gerontol 46(8): 694-702
Pubmed || Crossref || Others - 25. Hwang, J.W., Yao, H., Caito, S., et al. Redox regulation of SIRT1 in inflammation and cellular senescence. (2013) Free Radic Biol Med 61: 95-110.
Pubmed || Crossref || Others - 26. Park, E.J., Yi, J., Chung, K.H., et al. Oxidative stress and apoptosis induced by titanium dioxide nanoparticles in cultured BEAS-2B cells. (2008) Toxicol Lett 180(3): 222-229.
Pubmed || Crossref || Others - 27. Zhao, J., Bowman, L., Zhang, X., et al. Titanium dioxide (TiO2) nanoparticles induce JB6 cell apoptosis through activation of the caspase-8/Bid and mitochondrial pathways. (2009) J Toxicol Environ health A 72(19): 1141-1149.
Pubmed || Crossref || Others - 28. Yoo, K.C., Yoon, C.H., Kwon, D., et al. Titanium dioxide induces apoptotic cell death through reactive oxygen species-mediated Fas upregulation and Bax activation. (2012) Int J Nanomedicine 7: 1203-1214.
Pubmed || Crossref || Others - 29. Srivastava, R.K., Rahman, Q., Kashyap, M.P., et al. Nano-titanium dioxide induces genotoxicity and apoptosis in human lung cancer cell line, A549. (2013) Human & experimental Toxicol 32(2): 153-166.
Pubmed || Crossref || Others - 30. Lagopati, N., Tsilibary, E.P., Falaras, P., et al. Effect of nanostructured TiO(2) crystal phase on photoinduced apoptosis of breast cancer epithelial cells. (2014) Int J Nanomedicine 9: 3219-3230.
Pubmed || Crossref || Others - 31. Azim, S.A., Darwish, H.A., Rizk, M.Z., et al. Amelioration of titanium dioxide nanoparticles-induced liver injury in mice: Possible role of some antioxidants. (2015) Exper Toxicol Pathol 67(4): 305-314.
Pubmed || Crossref || Others - 32. Wang, Y., Cui, H., Zhou, J., et al. Cytotoxicity, DNA damage, and apoptosis induced by titanium dioxide nanoparticles in human non-small cell lung cancer A549 cells. (2015) Environ Sci pollut Research Int 22(7): 5519-5530.
Pubmed || Crossref || Others - 33. Park, E.J., Lee, S.Y., Lee, G.H., et al. Sheet-type titania, but not P25, induced paraptosis accompanying apoptosis in murine alveolar macrophage cells. (2014) Toxicology Letters 230(1): 69-79.
Pubmed || Crossref || Others - 34. Chen, J., Zhang, J., Cao, J., et al. Inflammatory MAPK and NF-κB signaling pathways differentiated hepatitis potential of two agglomerated titanium dioxide particles. (2016) J Hazard Mater 304: 370-378.
Pubmed || Crossref || Others - 35. Hu, H., Li, L., Guo, Q., et al. A mechanistic study to increase understanding of titanium dioxide nanoparticles-increased plasma glucose in mice. (2016) Food Chem Toxicol 95: 175-187.
Pubmed || Crossref || Others - 36. Childs, B.G., Baker, D.J., Kirkland, J.L., et al. Senescence and apoptosis: dueling or complementary cell fates? (2014) EMBO Rep 15(11): 1139-1153.
Pubmed || Crossref || Others - 37. Chen, H.W., Su, S.F., Chien, C.T. et al. Titanium dioxide nanoparticles induce emphysema-like lung injury in mice. (2006) FASEB J 20(13): 2393-2395.
Pubmed || Crossref || Others - 38. Gheshlaghi, Z.N., Riazi, G.H., Ahmadian, S., et al. Toxicity and interaction of titanium dioxide nanoparticles with microtubule protein. (2008) Acta Biochim Biophys Sin 40(9): 777-782.
Pubmed || Crossref || Others - 39. Hanot-Roy, M., Tubeuf, E., Guilbert, A., et al. Oxidative stress pathways involved in cytotoxicity and genotoxicity of titanium dioxide (TiO2) nanoparticles on cells constitutive of alveolo-capillary barrier in vitro. (2016) Toxicol in Vitro 33: 125-135.
Pubmed || Crossref || Others - 40. Wilhelmi, V., Fischer, U., Van Berlo, D., et al. Evaluation of apoptosis induced by nanoparticles and fine particles in RAW 264.7 macrophages: facts and artefacts. (2012) Toxicol in vitro 26(2): 323-334.
Pubmed || Crossref || Others - 41. Kang, S.J., Lee, Y.J., Kim, B.M., et al. Cytotoxicity and genotoxicity of titanium dioxide nanoparticles in UVA-irradiated normal peripheral blood lymphocytes. (2011) Drug and chemical Toxicol 34(3): 277-284.
Pubmed || Crossref || Others - 42. Bazak, R., Ressl, J., Raha, S., et al. Cytotoxicity and DNA cleavage with core-shell nanocomposites functionalized by a KH domain DNA binding peptide. (2013) Nanoscale 5(23): 11394-11399.
Pubmed || Crossref || Others - 43. Hou, Z., Zhang, Y., Deng, K., et al. UV-emitting upconversion-based TiO2 photosensitizing nanoplatform: near-infrared light mediated in vivo photodynamic therapy via mitochondria-involved apoptosis pathway. (2015) ACS Nano 9(3): 2584-2599.
Pubmed || Crossref || Others - 44. Pandurangan, M., Enkhtaivan, G., Young, J.A., et al. In Vitro Therapeutic Potential of Tio2 Nanoparticles Against Human Cervical Carcinoma Cells. (2016) Biol Trace Elem Res 171(2): 293-300.
Pubmed || Crossref || Others - 45. Ramkumar, K. M., Manjula, C., GnanaKumar, G., et al. Oxidative stress-mediated cytotoxicity and apoptosis induction by TiO2 nanofibers in HeLa cells. (2012) Eur J Pharm Biopharm 81(2): 324-333.
Pubmed || Crossref || Others - 46. Zhu, Y., Eaton, J.W., Li, C. Titanium dioxide (TiO2) nanoparticles preferentially induce cell death in transformed cells in a Bak/Bax-independent fashion. (2012) PLoS One 7(11): e50607.
Pubmed || Crossref || Others - 47. Kumar, S., Meena, R., Paulraj, R. Role of Macrophage (M1 and M2) in Titanium-Dioxide Nanoparticle-Induced Oxidative Stress and Inflammatory Response in Rat. (2016) Appl Biochem Biotechnol 180(7): 1257-1275.
Pubmed || Crossref || Others - 48. Kang, S.J., Kim, B.M., Lee, Y.J., et al. Titanium dioxide nanoparticles induce apoptosis through the JNK/p38-caspase-8-Bid pathway in phytohemagglutinin-stimulated human lymphocytes. (2009) Biochem Biophys Res Commun 386(4): 682-687.
Pubmed || Crossref || Others - 49. Kim, B.M., Lee, K.H., Hong, I.S., et al. p38 mitogen-activated protein kinase is a key regulator of 5-phenylselenyl- and 5-methylselenyl-methyl-2′-deoxyuridine-induced apoptosis in human HL-60 cells. (2012) Biochem Biophy Res commun 417(1): 237-244.
Pubmed || Crossref || Others - 50. Chandra, J., Orrenius, S. Mitochondria, oxygen metabolism and the regulation of cell death. (2002) Int Cong Series 1233: 259-272.
Pubmed || Crossref || Others - 51. Drakopanagiotakis, F., Xifteri, A., Polychronopoulos, V., et al. Apoptosis in lung injury and fibrosis. (2008) Eur Respir J 32(6): 1631-1638.
Pubmed || Crossref || Others - 52. Circu, M.L., Aw, T.Y. Reactive oxygen species, cellular redox systems, and apoptosis. (2010) Free Radic Biol Med 48(6): 749-762.
Pubmed || Crossref || Others - 53. Armand, L., Biola-Clier, M., Bobyk, L., et al. Molecular responses of alveolar epithelial A549 cells to chronic exposure to titanium dioxide nanoparticles: A proteomic view. (2016) J Proteomics 134: 163-173.
Pubmed || Crossref || Others - 54. Norman F.C. Cell Death. (2009) Ultrastructural Pathology the Comparative Cellular Basis of Disease: Wiley-Blackwell 32-59.
Pubmed || Crossref || Others - 55. Vaseva, A.V., Moll, U.M. The mitochondrial p53 pathway. (2009) Biochim Biophys Acta 1787(5): 414-420.
Pubmed || Crossref || Others - 56. Kroemer, G., Galluzzi, L., Brenner, C. Mitochondrial membrane permeabilization in cell death. (2007) Physiol Rev 87(1): 99-163.
Pubmed || Crossref || Others - 57. Norman F.C. Mitochondria. (2009) Ultrastructural Pathology the Comparative Cellular Basis of Disease: Wiley-Blackwell 181-198.
Pubmed || Crossref || Others - 58. Dasari, T.P., Pathakoti, K., Hwang, H.M. Determination of the mechanism of photoinduced toxicity of selected metal oxide nanoparticles (ZnO, CuO, Co3O4 and TiO2) to E. coli bacteria. (2013) J Environ Sci 25(5): 882-888.
Pubmed || Crossref || Others - 59. Sharma, V., Shukla, R.K., Saxena, N., et al. DNA damaging potential of zinc oxide nanoparticles in human epidermal cells. (2009) Toxicol Lett 185(3): 211-218.
Pubmed || Crossref || Others - 60. Hao, L., Wang, Z., Xing, B. Effect of sub-acute exposure to TiO2 nanoparticles on oxidative stress and histopathological changes in Juvenile Carp (Cyprinus carpio). (2009) J Environ Sci 21(10): 1459-1466.
Pubmed || Crossref || Others - 61. Ghosh, M., Bandyopadhyay, M., Mukherjee, A. Genotoxicity of titanium dioxide (TiO2) nanoparticles at two trophic levels: Plant and human lymphocytes. (2010) Chemosphere 81(10): 1253-1262.
Pubmed || Crossref || Others - 62. Yin, J.J., Liu, J., Ehrenshaft, M., et al. Phototoxicity of nano titanium dioxides in HaCaT keratinocytes—Generation of reactive oxygen species and cell damage. (2012) Toxicol Appl Pharma 263(1): 81-88.
Pubmed || Crossref || Others - 63. Zhong, H., Yin, H. Role of lipid peroxidation derived 4-hydroxynonenal (4-HNE) in cancer: Focusing on mitochondria. (2015) Redox Biol 4: 193-199.
Pubmed || Crossref || Others - 64. Huerta-García, E., Pérez-Arizti, J.A., Márquez-Ramírez, S.G., et al. Titanium dioxide nanoparticles induce strong oxidative stress and mitochondrial damage in glial cells. (2014) Free Radic Biol Med 73: 84-94.
Pubmed || Crossref || Others - 65. Chen, L., Zhou, L., Liu, Y., et al. Toxicological effects of nanometer titanium dioxide (nano-TiO2) on Chlamydomonas reinhardtii. (2012) Ecotoxicol Environ Saf 84: 155-162.
Pubmed || Crossref || Others - 66. Gurr, J.R., Wang, A.S., Chen, C.H., et al. Ultrafine titanium dioxide particles in the absence of photoactivation can induce oxidative damage to human bronchial epithelial cells. (2005) Toxicology 213(1–2): 66-73.
Pubmed || Crossref || Others - 67. Roy, R., Singh, S.K., Chauhan, L.K., et al. Zinc oxide nanoparticles induce apoptosis by enhancement of autophagy via PI3K/Akt/mTOR inhibition. (2014) Toxicol Lett 227(1): 29-40.
Pubmed || Crossref || Others - 68. 68. Zhang, Y., Yu, W., Jiang, X., et al. Analysis of the cytotoxicity of differentially sized titanium dioxide nanoparticles in murine MC3T3-E1 preosteoblasts. (2011) J Mater Sci Mater Med 22(8): 1933-1945.
Pubmed || Crossref || Others - 69. Dalai, S., Pakrashi, S., Joyce Nirmala, M., et al. Cytotoxicity of TiO2 nanoparticles and their detoxification in a freshwater system. (2013) Aquat Toxicol 138–139: 1-11.
Pubmed || Crossref || Others - 70. Kumari, J., Kumar, D., Mathur, A., et al. Cytotoxicity of TiO2 nanoparticles towards freshwater sediment microorganisms at low exposure concentrations. (2014) Environ Res 135: 333-345.
Pubmed || Crossref || Others - 71. Freyre-Fonseca, V., Delgado-Buenrostro, N.L., Gutiérrez-Cirlos, E.B., et al. Titanium dioxide nanoparticles impair lung mitochondrial function. (2011) Toxicol Lett 202(2): 111-9.
Pubmed || Crossref || Others - 72. Dalai, S., Pakrashi, S., Bhuvaneshwari, M., et al. Toxic effect of Cr(VI) in presence of n-TiO2 and n-Al2O3 particles towards freshwater microalgae. (2014) Aquat Toxicol 146: 28-37.
Pubmed || Crossref || Others - 73. Iverson, S.L., Orrenius, S. The cardiolipin–cytochrome c interaction and the mitochondrial regulation of apoptosis. (2004) Arch Biochem Biophys 423(1): 37-46.
Pubmed || Crossref || Others - 74. Li, N., Duan, Y., Hong, M., et al. Spleen injury and apoptotic pathway in mice caused by titanium dioxide nanoparticules. (2010) Toxicol Lett 195(2–3): 161-168.
Pubmed || Crossref || Others - 75. Paradies, G., Petrosillo, G., Pistolese, M., et al. The effect of reactive oxygen species generated from the mitochondrial electron transport chain on the cytochrome c oxidase activity and on the cardiolipin content in bovine heart submitochondrial particles. (2000) FEBS lett 466(2-3): 323-326.
Pubmed || Crossref || Others - 76. Periasamy, V.S., Athinarayanan, J., Al-Hadi, A.M., et al. Identification of titanium dioxide nanoparticles in food products: Induce intracellular oxidative stress mediated by TNF and CYP1A genes in human lung fibroblast cells. (2015) Environ Toxicol Pharmacol 39(1): 176-186.
Pubmed || Crossref || Others - 77. Boonrungsiman, S., Suchaoin, W., Chetprayoon, P., et al. Shape and surface properties of titanate nanomaterials influence differential cellular uptake behavior and biological responses in THP-1 cells. (2017) Biochem Biophys Reports 9: 203-210.
Pubmed || Crossref || Others - 78. Czajka, M., Sawicki, K., Sikorska, K., et al. Toxicity of titanium dioxide nanoparticles in central nervous system. (2015) Toxicol In Vitro 29(5): 1042-1052.
Pubmed || Crossref || Others - 79. Zhang, X., Li, W., Yang, Z. Toxicology of nanosized titanium dioxide: an update. (2015) Arch Toxicol 89(12): 2207-2217.
Pubmed || Crossref || Others - 80. Coppé, J.P., Desprez, P.Y., Krtolica, A., et al. The Senescence-Associated Secretory Phenotype: The dark Side of tumor Suppression. (2010) Annu Rev Pathol 5: 99-118.
Pubmed || Crossref || Others - 81. Sherr, C.J., Roberts, J.M. CDK inhibitors: positive and negative regulators of G1-phase progression. (1999) Genes Dev 13(12): 1501-1512.
Pubmed || Crossref || Others - 82. Firsanov, D.V., Solovjeva, L.V., Svetlova, M.P. H2AX phosphorylation at the sites of DNA double-strand breaks in cultivated mammalian cells and tissues. (2011) Clin Epigenetics 2(2): 283-297.
Pubmed || Crossref || Others - 83. Kuwano, K., Araya, J., Hara, H., et al. Cellular senescence and autophagy in the pathogenesis of chronic obstructive pulmonary disease (COPD) and idiopathic pulmonary fibrosis (IPF). (2016) Respir Investig 54(6):397-406.
Pubmed || Crossref || Others - 84. Dai, H., Deng, Y., Zhang, J., et al. PINK1/Parkin-mediated mitophagy alleviates chlorpyrifos-induced apoptosis in SH-SY5Y cells. (2015) Toxicology 334: 72-80.
Pubmed || Crossref || Others










