
Computational Approaches to Deciphering Binding Interactions among Tyrosinase and Urokinase-type Plasminogen Activator with Methimazole and Tranexamic Acid as Two Off-Label Antimelasmic Drugs
Arezou Lari1, Saeed Ivani2, Maryam Haghkhah1, Sara Ivani3
Affiliation
- 1Faculty of Pharmacy, Kermanshah University of Medical Sciences, Kermanshah, Islamic Republic of Iran
- 2Department of Biomedical Engineering and Medical Physics, ShahidBeheshti University of Medical Science, Tehran, Islamic Republic of Iran
- 3Department of Mathematics, Faculty of Science, Razi University, Kermanshah, Islamic Republic of Iran
- 4Department of Biology, Faculty of Science, Razi University, Kermanshah, Islamic Republic of Iran
Corresponding Author
Isaac Karimi; Department of Biology, Faculty of Science, Razi University, Kermanshah, Islamic Republic of Iran. E-mail: isaac_karimi2000@yahoo.com, karimiisaac@razi.ac.ir
Citation
Isaac Karimi., et al. Computational Approaches to Deciphering Binding Interactions among Tyrosinase and Urokinase- Type Plasminogen Activator with Methimazole and Tranexamic Acid as Two Off label Antimelasmic Drugs (2017) Bioinfo Proteom Img Anal 3(1):182- 186
Copy rights
© 2017 Isaac Karimi. This is an Open access article distributed under the terms of Creative Commons Attribution 4.0 International License.
Keywords
Melasma; Simulation; Molecular docking; Methimazole; Tranexamic acid
Abstract
Tyrosinase is rate-limiting enzyme for melanogenesis and urokinase-type plasminogen activator (uPA) plays an autocrine role in growth, differentiation and migration of keratinocyte involving in melanogenesis. Molecular dynamics simulations were performed to investigate the interaction of uPA and tyrosinase with methimazole (MIM) and tranexamic acid (TA) as two off-label antimelasmic drugs. Our findings showed that TA has more negative binding affinity to tyrosinase (-14.58 Kcal/mol) than that of uPA (-5.9 Kcal/mol) therefore its antimelasmic activity may be mediated through inhibition of tyrosinase with inhibition constant (IC) 20.5 pM rather than inhibition of uPA with IC 47.67 uM. Methimazole has approximately equal binding affinity to tyrosinase (-4.25 Kcal/mol) and uPA (-3.77 Kcal/mol) with IC 765.79 uM and 1.73 mM respectively. However MIM docked more stable with uPA. It seems that uPA plays more important role in antimelasmic activity of MIM. In conclusion, this in silico investigation prepared a theoretical framework that determines beneficial therapeutic effects of TA and MIM in hypermelanistic conditions like melasma.
Introduction
The outer layer of skin, epidermis, consists mainly of keratinocytes and melanocytes that their cooperation regulating skin pigmentation throughout a mutual chemical transportation of melanin, main integumentary protective pigment, and other chemical compounds. Tyrosinase (EC 1.14.18.1) is key copper-containing metalloenzyme in the melanogenesis that catalyzes the hydroxylation of tyrosine to L-3,4-dihydroxyphenylalanine (L-DOPA), the subsequent oxidation of L-DOPA to DOPA quinone and the oxidative conversion of 5,6-dihydroxyindole (DHI) and 5,6-dihydroxyindole-2-carboxylic acid (DHICA) to respective DHI-melanin and brown DHICA-melanin[1,2]. The DHI-melanin and DHICA-melanin then transformed and polymerized to dark eumelanin and brown pheomelanin as main skin pigments in skin melanocytes[3]. The melanin that produced in skin melanocytes will be transferred to skin keratinocytes via various commentary modes to regulate skin pigmentation[4]. Beside tyrosinase as a key regulator of pigmentation, urokinase-type plasminogen activator (uPA; EC3.4.21.73) is a serine protease that involved in skin pigmentation[5]. In this line, a large body of studies showed that uPA plays an autocrine role in growth, differentiation and migration of keratinocytes while it is produced by both melanocytes and keratinocytes[5]. In a pioneered work, Maeda and Tomita[6] showed that uPA generated by keratinocyte increases in vitro tyrosinase activity.
Melasma is an aesthetic hyperpigmentary disorder with poorly defined etiology mainly related to sun-exposure, genetic predisposition, hormonal imbalances during pregnancy, oral contraceptive intake, and phototoxic drugs[7,8]. The treatment modalities for melasma are frequently unsatisfactory due to exacerbation and remission and patients and dermatologists may feel discouraged even when melasma is treated with well-established formulations like pidobenzone[9]. Nevertheless, methimazole (MMI) and tranexamic acid (TA; Figure 1a & b) cocktail that prescribed as an “off-label” treatment has notable efficacy, well tolerated, and improved patient compliance, however its mode of action is not completely known.
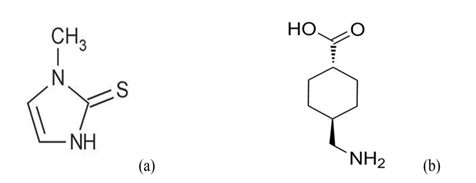
Figure 1: Chemical structures of methimazole (a) (1-methyl-3H-imidazole-2-thione; (b) tranexamic acid (trans-4-(amino methyl) cyclohexanecarboxylic acid.
In this continuum, MMI as a potent peroxidase inhibitor, is a classic antithyroid drug which has been recently discovered to have a depigmenting effect when topically prescribed in guinea pigs and human[10]. Histologically, topical applications of MMI significantly reduced the melanin content of the epidermis and melanocytes exhibited morphological changes without a significant decrease in their number[10]. In a mechanistic investigation, TA inhibited melanin synthesis in melanocytes by interfering with the interaction of melanocytes and keratinocytes through inhibition of the plasminogen/plasmin system[6]. This synthetic lysine derivative is a competitive inhibitor of plasminogen that prevents the breakdown of fibrin[11,12]. Its haemostatic property is limited to its antifibrinolytic effect and it does not interfere with the other blood-clotting parameters[13,14]. Hence molecular mechanic investigation of tyrosinase and uPA with two putative inhibitors (TA and MMI) has been focus of this study.
Materials and Methods
Data preparation: The X-ray crystal structures of Bacillus megaterium tyrosinase (PDB ID 4P6S) and Homosapiens urokinase-type plasminogen activator (PDB ID 3KGP) were retrieved from protein data bank (PDB) at the Research Collaboratory for Structural Bioinformatics (http://www.RCSB.org)[15]. Protein preparations like repairing side chains, adding hydrogen atoms and other modifications needed for docking were performed by using Chimera and Molegro software[16]. The 3D structures of ligands were constructed using the Sketch Molecule and were fully minimized through applying MM+ and AM1 in HyperChem package[17].
Molecular docking: Molecular docking studies were used to explore the binding affinities and interaction modes between protein and ligand using Auto dock Tools-1.5.6[18]. The ligand–protein complexes were analyzed to identify the interactions, binding affinity and inhibition constant. Visual Molecular Dynamics (VMD1.9.1)[19] and LigPlot+[20] softwares were used as graphical interface to analyze the simulation results. The binding site was defined as a spherical region which encompasses all protein atoms within 10.0Å of each crystallographic ligand atoms. Default settings were used for all calculations.
The docking scores were recorded and the best docking poses were selected based on the more favorable binding energy and interactions between ligand and protein. Structures of the best conformers were used as starting point for molecular dynamics (MD) simulations.
MD simulation: In order to investigate and track the behavior of aforementioned medicines into the active or binding sites of both proteins, MD simulation was utilized using GROMACS 4.5.3 package[21]. In this study, two independent MD simulations were set up including complexes of protein and ligand.
The topology files for the ligands were generated by Automated Topology Builder (ATB version 2.2[22]. Protein systems were solvated in a cubic box with simple point charge water model at “10.0 Å marginal radii” uses. The structures were found to be positively charged at physiological pH 7.4, therefore two chlorides (Cl-) were added to the simulation box to make the system electrically neutral.
Initially the solvent molecules were relaxed while all the solute atoms were harmonically restrained to their original positions with a force constant of 100 kcal/mol for 5,000 steps. After this, whole molecular systems were subjected to energy minimization for 5,000 iterations using steepest descent algorithm implementing GROMOS96 43a1 force field. Berendsen temperature coupling method was used to regulate the temperature inside the box[23]. Electrostatic interactions were computed using the particle mesh Ewald method[24]. The pressure was maintained at 1.0 atm with the allowed compressibility range of 4.5 × 10-5 atm. The linear constraint solver algorithm was employed to constrain bond lengths involving hydrogen atoms, permitting a time step of 2 fs[25] while Van der Waals and coulomb interactions were truncated at 1.0 nm. The non-bonded pair list was updated every 10 steps and conformations were stored every 0.5 ps. The systems were subjected to MD simulation for 20 ns. Then root mean square deviation (RMSD), root mean squared fluctuation (RMSF), solvent accessible surface area (SASA) and radius of gyration (Rg) analysis were calculated to predict dynamic structural transformations for studied enzymes.
Results and Discussion
Computational methods were employed to study the interaction of candidate enzymes, tyrosinase and uPA, involving melanogenesis with TA and MIM. Active sites of the proteins were known from the co-crystallized ligands from PDB files. After docking, the best conformers with the lowest binding energy were collected and used for further investigations. The results obtained from docking analysis further motivated us to study the dynamic behavior of both enzymes (vide infra).
The TA and MMI were docked into the binding sites of tyrosinase and uPA (see Figures. 2a & b, 3a & b). Auto dock provided six conformations of TA with tyrosinase with binding affinity range of -14.30 to -14.58 in clusters while docking results of TA with uPA led to only one cluster with a binding affinity range of -5.83 to -5.90 (Table 1).
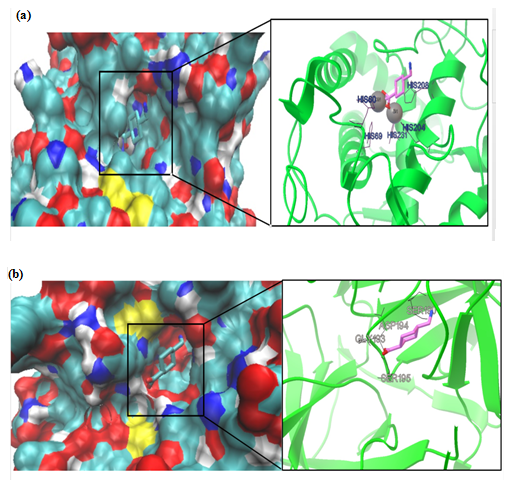
Figure 2: Molecular docking simulations of binding between tyrosinase (PDB ID 4P6S; (a) or urokinase-type plasminogen activator (PDB ID 3KGP; (b) and tranexamic acid are depicted. The left photos corresponding surface structure of protein interacting with tranexamic acid. The cyanic parts show the residues of protein, which bind with tranexamic acid to develop the catalytic core domain. The coarser stick structure in blue was used to represent tranexamic acid, which is situated in the catalytic core domain. The interactions between tranexamic acid and protein are shown in right photos. The coarser stick structure mainly in purple on the right side was used to represent the tranexamic acid while the coarser stick with black colors shows the residues of proteins.
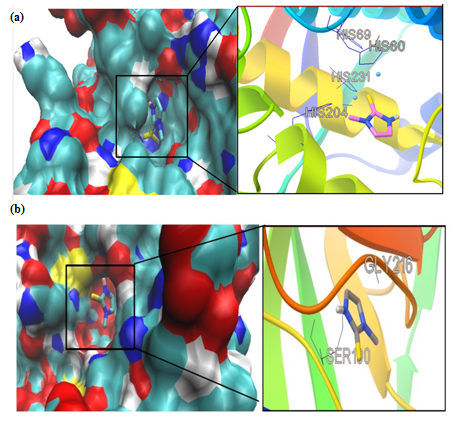
Figure 3: Molecular docking simulations of binding between tyrosinase (PDB ID 4P6S); (a) or urokinase-type plasminogen activator (PDB ID 3KGP); (b) and methimazole are depicted. The left photos corresponding surface structure of protein interacting with methimazole. The cyanic parts show the residues of protein, which bind with methimazole to develop the catalytic core domain. The coarser stick structure in blue was used to represent methimazole which is situated in the catalytic core domain. The interactions between methimazole and protein are shown in right photos. The coarser stick structure mainly in gray on the right side was used to represent the methimazole while the coarser stick with black shows the residues of proteins.
Table 1: Molecular docking of binding between and methimazole (MIM) and tranexamic acid (TA) in the active sites of tyrosinase (PDB ID 4P6S) or urokinase-type plasminogen activator (uPA; PDB ID 3KGP).
| Ligand-protein | Conformers no. in cluster/total conformers no. | Inhibition constant | Binding affinity (Kcal/mol) |
|---|---|---|---|
| TA-tyrosinase | 6/10 | 20.50pM | -14.58 |
| TA-uPA | 10/10 | 47.67 uM | -5.90 |
| MIM-tyrosinase | 9/10 | 765.79 uM | -4.25 |
| MIM-uPA | 10/10 | 1.73 mM | -3.77 |
All conformers of TA complex with uPA made hydrogen bonding with ASP194, GLY193, SER190 and SER195 in active site as confirmed by re-docking (Figure 2a & b). Our findings clearly showed that TA has more negative binding affinity to tyrosinase rather than uPA therefore it is more favorable to ascribe its antimelasmic activity by inhibition of tyrosinase (Table 1). Moreover the inhibition constant (IC50) that declares the concentration required to produce half maximum inhibition of enzymes was lower for TA to inhibit tyrosinase rather than uPA (Table 1). In this regard, TA has shown to provide rapid and sustained lightening in melasma by decreasing melanogenesis in epidermal melanocytes[26]. However, the in vitro culture of melanocyte in the presence of keratinocyte-conditioned medium strongly suggested uPA-inhibitory role of TA in melanogenesis[6]. The IC50 of TA for inhibiting uPA shows that TA has acceptable inhibitory effect on uPA (Table 1). We performed MD simulation on double chain uPA while experimental studies employed single chain one[6,27]. Plasmin and plasmin activators like uPA are serine proteases and antifibrinolytic activity of TA is mediated by its strong affinity for the five lysine-binding sites of plasminogen and its competitive inhibition of the conversion of plasminogen to plasmin[28] or noncompetitive inhibition of plasmin at higher concentration[29].
In this study we used substrate-free crystal structure of tyrosinase from Bacillus megaterium (TyrBm) as tyrosinase model[30]. In TyrBm, the Cu2+ has been replaced by Zn2+ ions without structural consequences, while the presence of Zn2+ ions inhibited the activity of tyrosinase on both monophenols and diphenols as substrates[31,32]. All conformers of TA complex with tyrosinase made hydrogen bonding with HIS residues (HIS60, HIS69, HIS204, HIS208, and HIS231) in active site as described previously in crystal structure of TyrBm[30] (Figure 2a & b). Therefore, we hypothesize that TA would be a competitive inhibitor of tyrosinase and competes against monophenol (tyrosine) or diphenol (L–DOPA) substrates[30]. Recently, results of an in vitro study showed that TA-treated melanocytes exhibited reduced melanin content and tyrosinase activity and TA also decreased tyrosinase, tyrosinase-related protein (TRP)-1, and TRP-2 protein levels[33]. Our study is the first one that theoretically supports molecular mechanics of inhibitory effect of TA on melanogenesis through inhibition of tyrosinase.
We had only one cluster for all 10 conformations in MMI docking with proteins (Table 1). The binding affinity was -4.25 and -3.77 in complex of MMI with tyrosinase and uPA, respectively (Figure 3a & b). After docking, conformers with the lowest binding energy were screened and used for further investigations. The RMSD for all Cα atoms from the initial structures were examined. The magnitude of fluctuations in both enzymes showed very small difference in average RMSD values after the relaxation period (Figure 4a & b). This leads to produce stable trajectories in simulation. We observed that MIM-bound tyrosinase was so mobile while MIM-bound uPA followed a stable dynamics (Figure 4a & b). In addition, RMSF of individual backbone atoms of residues were calculated along the simulation for each enzyme inhibitor complex. The results revealed the stability of amino acids (Figure 5a & b). In sum, we have found that structural stability of tyrosinase complexes were declined in most of the domains for both medicines especially MIM.
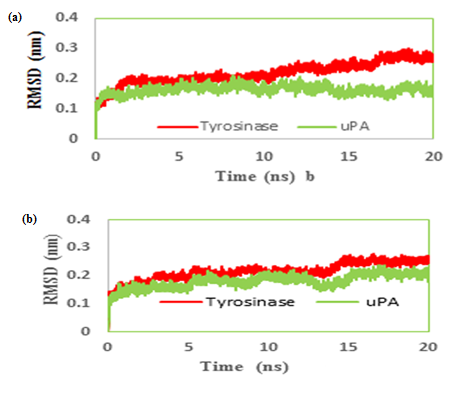
Figure 4: Root mean square deviation (RMSD) evolution of carbon alpha of (a) tranexamic acid (b) methimazole; bound to urokinase-type plasminogen activator (uPA) and tyrosinase during 20 ns MD simulations.
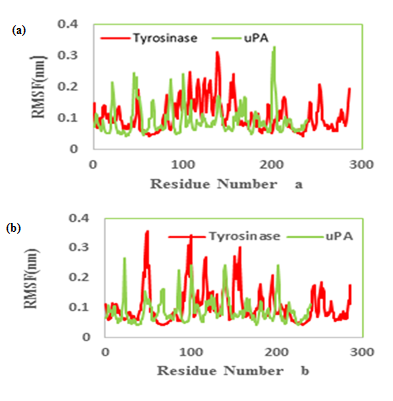
Figure 5: Molecular dynamics root mean squared fluctuation (RMSF) values of the active site carbon alpha of (a) tranexamic acid (b) methimazole bound to urokinase-type plasminogen activator (uPA) and tyrosinase.
The radius of gyration (Rg) is defined as the massweight root meansquare distance of collection of atoms from their common center of mass. Therefore it provides an insight into the overall dimension of the protein. The Rg plot for Cα atoms of protein is shown (Figure 6a & b) and based on its results both simulated systems have been stabilized during MD simulation
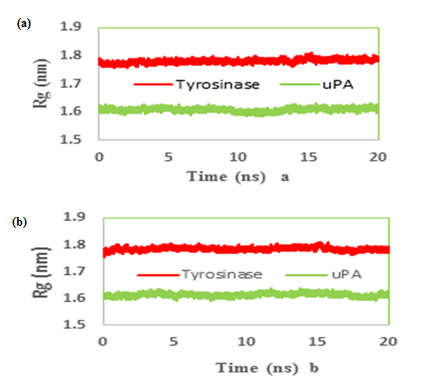
Figure 6: Time evolution of the radius of gyration (Rg) during 20 ns of MD simulation of (a) tranexamic acid (b) methimazole bound to urokinase-type plasminogen activator (uPA) and tyrosinase.
We also reported the number of hydrogen bonds between the TA and MIM (Figure 7a & b) bounded to uPA and tyrosinase during the MD simulation. In the complex of TAuPA, significant H-bondings have been observed between ligand and SER (48.70%), TYR (45.42%) and GLY (30.00%) residues during simulation. For docking TA with tyrosinase, H-bondings were scarce. In this line, less than 4 percent of all H-bondings interact between N1 atom of ligand as a donor with ASN205, MET215 and GLY216 residues of tyrosinase as acceptors. In MIM complex simulations, the amounts of H-bonding were less than 3 percent, however the interacting residues were GLY216, GLN192 in uPA and GLY213, ARG209, ASN152, LYS150 in tyrosinase. The alterations of SASA of tyrosinase and uPA during MD simulation are shown (Figure 8a & b) and their stable trends validate the accuracy of MD simulation.
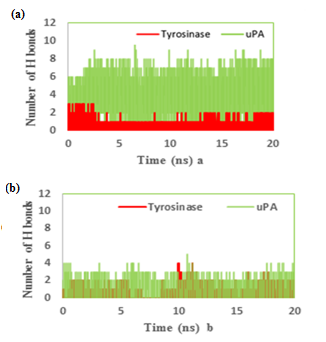
Figure 7: The number of hydrogen bonds between the (a) tranexamic acid or (b) methimazole bound to urokinase-type plasminogen activator (uPA) and tyrosinase during the MD simulation.
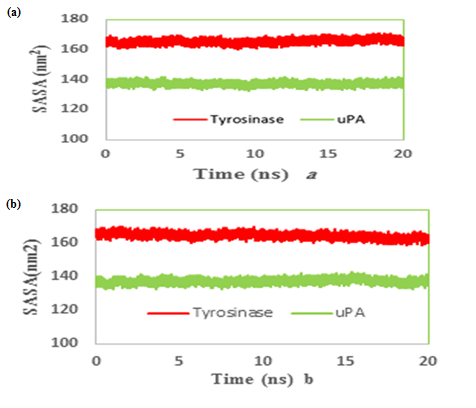
Figure 8: Time evolution of the total solvent accessible surface (SASA) during 20 ns of MD simulation of (a) tranexamic acid or (b) methimazole bound to urokinase-type plasminogen activator (uPA) and tyrosinase.
Conclusion
In sum, our in silico findings showed possible therapeutic mechanism of TA and MIM in melasma. Here we showed that TA has more negative binding affinity and lower inhibition constant to tyrosinase rather than uPA therefore it is more encouraging to ascribe its antimelasmic activity through interacting with tyrosinase. MIM inhibited tyrosinase and uPA approximately in a same amount however it docked more stable with uPA than tyrosinase. It seems that tyrosinase also plays more important role in the antimelasmic activity of MIM.
Disclosures:
The authors declared no conflicts of interest.
References
- 1. Espín, J.C., Varón, R., Fenoll, L.G., et al. Kinetic characterization of the substrate specificity and mechanism of mushroom tyrosinase. (2000) Eur J Biochem 267(5): 1270–1279.
- 2. Raynova, Y., Doumanova, L., Idakieva, K.N. Phenoloxidase activity of Helix aspersamaxima (garden snail, gastropod) hemocyanin. (2013) Protein J 32: 609–618.
- 3. Rodríguez-López, J.N., Tudela, J., Varón, R., et al. Analysis of a kinetic model for melanin biosynthesis pathway. (1992) J Biol Chem 267(6): 3801–3810.
- 4. Delevove, C. Melanin transfer: the keratinocytes are more than gluttons. (2014) J Invest Dermatol 134(4): 877–879
- 5. Jensen, P.J., John, M., Baird, J. Urokinase and tissue type plasminogen activators in human keratinocyte culture. (1990) Exp Cell Res 187(1): 162–169.
- 6. Maeda, K., Tomita, Y. Mechanism of the inhibitory effect of tranexamic acid on melanogenesis in cultured human melanocytes in the presence of keratinocyte-conditioned medium. (2007) J Health Sci 53(4): 389–396.
- 7. Rendon, M., Berneburg, M., Arellano, I., et al. Treatment of melasma. (2009) J Am Acad Dermatol 54: 272–281.
- 8. Arora, P., Sarkar, R., Garg, V.K., et al. Lasers for treatment of melasma and post-inflammatory hyper pigmentation. (2012) J Cutan Aesthet Surg 5(2): 93–103.
- 9. Zaniri, F., Asad, G.B., Campolmi, P., et al. Melasma: successful treatment with pidobenzone 4%. (2008) Dermatol Ther 21: 18–19.
- 10. Malek, J., Chedraoui, A., Nikolic, D., et al. Successful treatment of hydroquinone – resistant melasma using topical methimazole. (2013) Dermatol Ther 26(1): 69–72.
- 11. Dang, P., Schwarzkopf, R. Tranexamic acid and total knee arthroplasty. (2013) Ann OrthopRheumatol 714: 5456–5759.
- 12. Wong, J., Abrishami, A., El Beheiry, H., et al. Topical application of tranexamic acid reduces postoperative blood loss in total knee arthroplasty: a randomized, controlled trial. (2010) J Bone Joint Surg Am 92(15): 2503–2513.
- 13. Piamphongsant, T. Treatment of melasma: A review with personal experience. (1998) Int J Dermatol 37(12): 897–903.
- 14. Khan, M.F., Khan, M.F., Ashfaq, M., et al. Comparative evaluation of some new tranexamic acid derivatives and their copper (II) complexes for anti-tumor, analgesic, bactericidal and fungicidal activities. (2002) Pak J Pharm Sci 15(1): 55–62.
- 15. Berman, H.M., Westbrook, J., Feng, Z., et al. The protein data bank. (2000) Nucleic Acids Res 28(1): 235–242.
- 16. Pettersen, E.F., Goddard, T.D., Huang, C.C., et al. UCSF Chimera-a visualization system for exploratory research and analysis. (2004) J Comput Chem 25(13): 1605–1612.
- 17. HyperChem (TM) Professional 7.51, Hypercube, Inc., 1115 NW 4th Street, Gainesville, Florida 32601, USA.
- 18. Morris, G.M., Huey, R., Lindstrom, W., et al. Autodock4 and AutoDockTools4: automated docking with selective receptor flexibility. (2009) J Comput Chem 30(16): 2785–2791.
- 19. Humphrey, W., Dalkem A., Schulten, K. VMD: Visual molecular dynamics. (1996) J Mol Graph 14(1): 33–38.
- 20. Laskowski, R.A., Swindells, M.B. LigPlot+: multiple ligand–protein interaction diagrams for drug discovery. (2011) J Chem Inf Model 51(10): 2778–2786.
- 21. Hess, B., Kutznerm C., van der Spoel, D., et al. GRGMACS 4: algorithms for highly efficient load-balanced and scalable molecular simulation. (2008) J Chem Theory Comput 4(3): 435–447.
- 22. Malde, A.K, Zuo, L., Breeze, M., et al. An automated force field topology builder (ATB) and repository: version 1.0. (2011) J Chem Theory Comput 7(12): 4026–4037.
- 23. Berendsen, H.J.C., Postma, J.P.M., van Gunsteren, W.F., et al. Molecular-dynamics with coupling to an external bath. (1984) J Chem Phys 81(8): 3684–3690.
- 24. Cheatham, T.E., Miller, J.L., Fox, T., et al. Molecular dynamics simulations on solvated biomolecular systems: the particle mesh Ewald method leads to stable trajectories of DNA RNA and proteins. (1995) J Am ChemSoc 117(14): 4193–4194.
- 25. Hess, B., Bekker, H., Berendsen, H.J.C. LINCS: A linear constraint solver for molecular simulations. (1997) J Comp Chem 18(12): 1463–1472.
- 26. Karn, D., Kc, S., Amatya, A., et al. Oral tranexamic acid for the treatment of melasma. (2012) Kathmandu Univ Med J 10(4): 40–43.
- 27. Takada, A., Takada, Y. Inhibition by tranexamic acid of the conversion of single-chain tissue plasminogen activator to its two chain form by plasmin: the presence on tissue plasminogen activator of a site to bind with lysine binding sites of plasmin. (1989) Thromb Res 55(6): 717–725.
- 28. K?ak, M., Anäkkälä, N., Wang, W., et al. Tranexamic acid, an inhibitor of plasminogen activation, aggravates staphylococcal septic arthritis and sepsis. (2010) Scand J Infect Dis 42(5): 351–358.
- 29. Reichel, C.A., Lerchenberger, M., Uhl, B., et al. Plasmin inhibitors prevent leukocyte accumulation and remodeling events in the post ischemic microvasculature. (2011) PLoS One 6(2): e17229.
- 30. Goldfeder, M., Kanteev, M., Isaschar-Ovdat, S., et al. Determination of tyrosinase substrate-binding modes reveals mechanistic differences between type-3 copper proteins. (2014) Nat Commun 5: 4505–4505.
- 31. Han, H.Y., Zou, H.C., Jeon, J.Y., et al. The inhibition kinetics and thermodynamic changes of tyrosinase via the zinc ion. (2007) Biochim Biophys Acta 1774(7): 822–827.
- 32. Sendovski, M., Kanteev, M., Shuster Ben-Yosef, V., et al. First structures of an active bacterial tyrosinase reveal copper plasticity. (2011) J Mol Biol 405(1): 227–237.
- 33. Kim, M.S., Bang, S.H., Kim, J.H., et al. Tranexamic acid diminishes laser-induced melanogenesis. (2015) Ann Dermatol 27(3): 250–256.










