
Development of Therapy-Related Myeloid Neoplasms in the Pediatric Population Post Chemotherapy for Ewing Sarcoma
Rachel E. Goff1, Michella K. Whisman2, Dragon C. Do3, Joana M. Mack3, and Rebecca A. Levy2,4*
Affiliation
1University of Arkansas for Medical Sciences, College of Medicine, Little Rock, AR
2University of Arkansas for Medical Sciences, Department of Pathology, Little Rock, AR
3Arkansas Children’s Hospital, Department of Hematology and Oncology Little Rock, AR
4Arkansas Children’s Hospital, Department of Pathology, Little Rock, AR
Corresponding Author
Rebecca A. Levy, MD, Assistant Professor, University of Arkansas for Medical Sciences, Department of Pathology, 4301 West Markham Street, Slot 517, Little Rock, Arkansas 72205, Tel: (501) 686-7966/ Fax: (501) 603-1479; E-mail: ralevy@uams.edu
Citation
Levy, R.A., et al. Development of Therapy-Related Myeloid Neoplasms in the Pediatric Population Post Chemotherapy for Ewing Sarcoma. (2018) Int J Cancer Oncol 5(1): 41- 45.
Copy rights
© 2018 Levy, R.A. This is an Open access article distributed under the terms of Creative Commons Attribution 4.0 International License.
Keywords
Ewing Sarcoma; Neoplasms; Pediatrics; Chemotherapy
Abstract
Therapy-related myeloid neoplasms (t-MN) are a rare side effect of antineoplastic therapy including radiation and certain chemotherapeutic agents. Risk of t-MN is generally correlated with the specific agent and dosage used. Although rare, these disorders typically occur 5-7 years after therapy with alkylating agents and 1-3 years after therapy with topoisomerase II inhibitors. In the past decade, we have two pediatric patients who developed t-MN within a year of completing treatment for Ewing sarcoma. Both patients began treatment within a month of their primary diagnosis, which involved a regimen of radiation, alkylating agents, and topoisomerase II inhibitors. They each experienced complications of delayed count recovery, requiring use of granulocyte-colony stimulating factor. At her six-month follow-up, one patient was noted to have hypocellular marrow with increased blast population, consistent with therapy-related acute myeloid leukemia. The second patient was diagnosed with hypocellular marrow for age with trilineage dyspoiesis and increased blasts (10-15%), consistent with therapy-related myelodysplastic syndrome at her one-year follow-up appointment. In general, both patients were doing well post chemotherapy, with minimal complaints of fatigue and awaiting blood cell count recovery. Notably, the latency period for therapy related myeloid neoplasms may overlap with the expected waiting period for marrow recovery- particularly for topoisomerase II inhibitors such as doxorubicin and etoposide. Our case study suggests it is prudent to research this phenomenon thoroughly and perform a bone marrow core biopsy to exclude the possibility of rapidly evolving t-MN in patients with slow count recovery.
Introduction
The Ewing sarcoma (ES) family of tumors is a group of small, round-cell neoplasms of neuroectodermal origin arising in bone or soft tissue[1] and commonly occurs in the pediatric to young adult population (5 - 25 years old).Nearly all cases of ES have the classic translocation t(11;22)(q24;q12) that encodes the EWSR1/FLI-1 fusion transcript[2]. Prognosis is affected by whether the tumor occurs as a local lesion or presents with metastasis at the time of diagnosis and if the tumor has a good pathologic response to therapy[3]. Currently, in patients with localized ES, the expected event free survival rate at five years is around 70%[4].
Treatment algorithms can include induction chemotherapy, local treatment including surgical intervention, maintenance chemotherapy, and consolidation high-dose chemotherapy (HDCT) based on alkylating agents. The overall survival rate of Ewing sarcoma in the early 1970s was reported to be < 10%[5]. The addition of the alkylating agent ifosfamide and topoisomerase II inhibitor etoposide to investigational treatment algorithms in the 1980s showed significantly improved outcomes for patients with nonmetastatic Ewing sarcoma and became a part of the standard chemotherapy regimen in the early 1990s[6,7]. The addition of ifosfamide and etoposide did not improve survival or outcomes in patients with metastases[4]. The addition of these medications has been associated with increased rates of secondary malignancies[6,8] in patients with Ewing sarcoma. Secondary hematopoietic malignancies typically develop within the first few years (eg 1 - 5 years) after treatment and are associated with chemotherapy Regimen, while secondary solid tumors (sarcoma and carcinoma) occur later with a typical latent period of 10 years or more and are strongly associated with radiation[9-11]. Children’s Oncology Group (COG) conducted a large study, INT-0091, evaluating secondary malignancies in patients with nonmetastatic Ewing tumor Treatment; the COG identified 2% t-MNs at 5 years[6,12] whereas a European study identified less than 1% t-MNs at 5 years[13]. In the same COG trial (INT-0091), patients with metastatic ES, treated with higher doses of chemotherapy (including cyclophosphamide, ifosfamide and doxorubicin) and G-CSF, the incidence of secondary t-MN (leukemia and MDS) increased to 11% at 5 years without improving event free survival or overall survival[6,14].
The World Health Organization Tumors of Haematopoietic and Lymphoid Tissue define therapy-related Myelodysplastic Syndrome (t-MDS) and therapy-related Acute Myeloid Leukemia (t-AML) as a late complication of cytotoxic chemotherapy or radiation therapy administered for a prior neoplastic or non-neoplastic disorder[15]. The latency period between the primary disease diagnosis and t-MN onset ranges from several months to years and is associated with the cumulative dose, dose intensity, and type of preceding cytotoxic therapy[16]. In patients treated with alkylating agents or radiotherapy, bone marrow myelodysplastic changes usually precedes t-AML allowing for a longer latency period of 5-7 years and are more likely to have complex karyotypes[17,18]. Patients previously treated with topoisomerase II inhibitors usually present with balanced translocations, have a shorter latency period (1 - 3 years), and exhibit a rapidly progressive leukemia[17,19]. Therapy related MDS in the setting of topoisomerase II inhibitors is commonly defined by balanced translocations with 11q23 and 21q22[16-18,20-22]. Studies have shown that a portion of young patients with t-MNs can experience long term survival with allogenic stem cell transplantation[16]. Outcomes of post-transplant patients are affected by transplant related complications, non-complete remission/relapse, and poor cytogenetics[1,23,24].
Case Presentation
We will discuss 2 cases of Ewing sarcoma with rapid therapy related MDS/therapy related AML.
Patient 1 was a 17 year old female who presented after several months of worsening left hip pain. Physical exam was notable for fullness and tenderness inferior to the left iliac wing. Imaging revealed a lytic lesion along the left iliac wing with extension into the left iliacus and gluteus muscles. Her metastatic workup was negative. A CT-guided core biopsy of the left iliac wing was performed by interventional radiology. The histologic sections showed diffuse tumor involvement which was composed of monomorphic, small round blue cells with scant cytoplasm and clear chromatin. There was diffuse membranous staining with CD99 and molecular studies revealed a EWSR1/FLI1 fusion transcript (see Table 2). The constellation of these findings is diagnostic of Ewing sarcoma.
Patient 1 was enrolled onto COG Study AEWS1031, Regimen B, which consists of 3 different combinations of chemotherapy including vincristine-topotecan-cyclophosphamide, ifosfamide-etoposide, and vincristine-doxorubicin-cyclophosphamide along with local radiation therapy. Pegfilgrastim was utilized routinely during treatment. She finished treatment after one year. Her end of therapy evaluation revealed no evidence of recurrent or progressive disease, confirming that she was in remission.
About 6 months later (see Table 1) she began having headaches and fatigue. Her physical exam was unremarkable aside from conjunctival pallor. While her CBCs following end of therapy had not shown full count recovery yet, the CBC this visit was markedly abnormal with pancytopenia (white blood cell count 1.28 K/uL, hemoglobin 6.3 g/dL, platelet 114 K/uL, and ANC 280). The white blood cell differential was otherwise unremarkable. A bone marrow biopsy was performed and demonstrated a hypocellular bone marrow for age (20% cellularity) with an increased number of CD34-positive cells (see Figure 1). In total, the CD34-positive blasts accounted for 50-60% of the total nucleated cells by immunohistochemical stain. On the accompanying aspirate, intermediate-sized blasts with immature chromatin, prominent nucleoli, and scant to moderate cytoplasm were identified. Flow cytometry on the bone marrow aspirate revealed 33% blasts, confirming the diagnosis of t-AML. Additional ancillary studies revealed a 3q26.2 gain in 55% of cells, a 7cen 7q31 deletion/monosomy in 68% of cells, and a 22q22 (RUNX1) gain in 80.5% of cells by fluorescent in situ hybridization (FISH) (see Table 2).
Table 1: Time line of diagnoses and treatment.
| Patient | Symptoms began | Diagnosis of Ewing’s | Treatment began | End of treatment | Secondary diagnosis | Start of Secondary treatment | Current status |
|---|---|---|---|---|---|---|---|
| 1 | 6/2014 | 3/2015 | 3/2015 | 3/2016 | 10/2016 | 10/2016 | Stem cell transplant 2/2017, 1 year in remission |
| 2 | 5/2014 | 8/2014 | 9/2014 | 11/2015 | 12/2016 | 12/2016 | Progression to AML; Expired 8/2017 |
Table 2: Cytogenetic/Molecular findings.
| Patient 1 | Patient 2 | |
|---|---|---|
| Original Diagnosis | Ewing Sarcoma | Ewing sarcoma/primitive neuroectodermal tumor |
| Location | Left iliac wing | Skull and dura mater |
| Age at diagnosis | 17 | 11 |
| Molecular | EWSR1/FLI1 detected by RT-PCR amplification; Consistent with type I fusion transcript | EWSR1/FLI1 detected by RT-PCR amplification; Consistent with type II fusion transcript |
| Secondary Diagnosis | Therapy related myeloid neoplasm | Therapy related myeloid neoplasm |
| Specific type | t-AML | t-MDS |
| Cytogenetics | 46, XX, t(3;8)(q26;q24), -7, +mar [1]; 46, sl, (12;21)(q12;q22), [16]; 46, XX [3] | 45, XX, add(3)(q27), -7 [20] |
| Fluorescence in situ hybridization | 3q26.2 (MECOM) gain in 55% 7cen(D7Z1) 7q31(D7S486) deletion/monosomy in 68% 21q22 (RUNX1) gain in 80.5% | 7cen(D7Z1) 7q31(D7S486) deletion/monosomy in 81% |
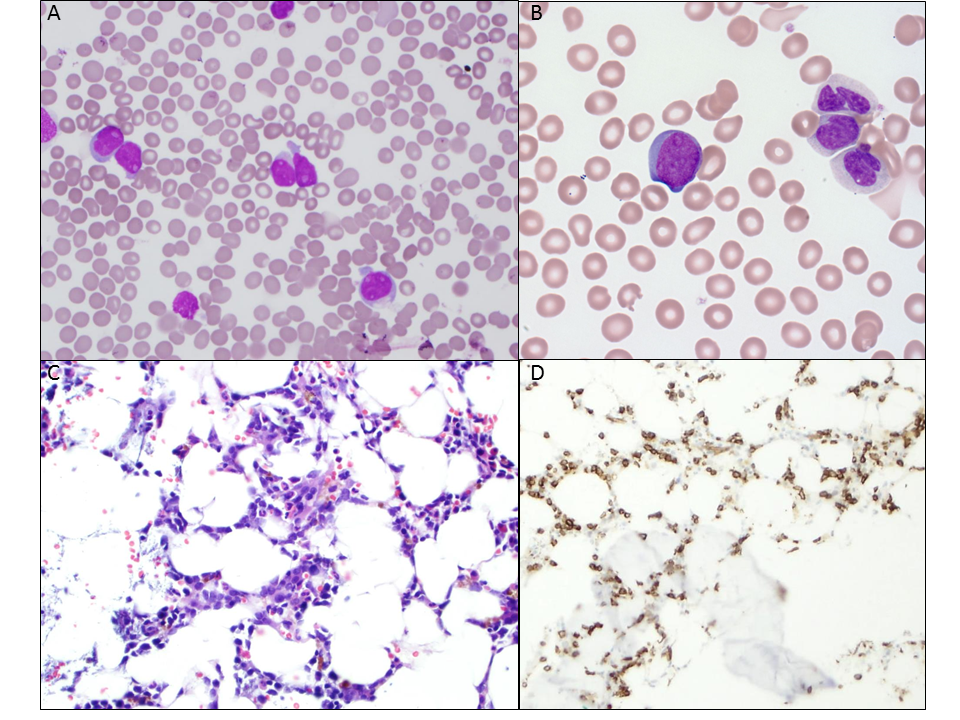
Figure 1: Patient 1 Bone Marrow Findings. A) Aspirate smear (400x) demonstrating increased and prominent blasts. B) Blasts identified circulating in the peripheral blood (1000x). C) H&E of the bone marrow biopsy (200x) showing numerous large cells with a high nuclear: cytoplasm ratio amidst maturing granulocytes. D) CD34 immunostain of the bone marrow biopsy (200x) delineates many of the large cells as blasts.
She began salvage therapy with high dose cytarabine and mitoxantrone-dexrazoxane. She developed invasive fungal sinusitis with Curvularia lunata for which she received treatment with liposomal amphotericin B. She also received decitabine and gemtuzuma bozogamicin with eventual bone marrow minimal residual disease (MRD) testing revealing 4.4% blasts by flow cytometry. She then proceeded to a matched unrelated donor peripheral blood stem cell transplant, conditioned with fludarabine-busulfan-rabbit ATG. Her bone marrow chimerism study at Day +100 showed 100% donor cells. She did not have complications of graft-versus-host disease (GVHD) and continued to do well at her Day +112 clinic visit (see table 1).
Patient 2 was a 10 year old female who presented after having 2 - 3 months of worsening right sided frontal/temporal headaches, associated with right sided pain/weakness and intermittent vision loss. Her physical exam was notable for tremors along bilateral upper extremities. An MRI of the brain revealed an extra-axial dural-based mass in the right temporal convexity with associated midline shift. A skull and dura mater resection of the mass was performed by neurosurgery which showed a high-grade small round blue cell neoplasm. The neoplasm demonstrated sheets of cells with primitive features, including inconspicuous cytoplasm, open chromatin, and prominent nucleoli, amidst scattered necrosis and hemorrhage. CD99 showed diffuse expression. Real time-Polymerase chain reaction (RT-PCR) identified an EWSR1/FLI1 fusion transcript (see Table 2). The final diagnosis was a Ewing sarcoma/primitive neuroectodermal tumor.
Her workup, including pan CT, bone scan, and bone marrow evaluation, was negative for metastatic disease. She began standard therapy with vincristine-doxorubicin-cyclosphosphamide and ifosfamide-etoposide. Pegfilgrastim was utilized routinely during treatment. She received radiation therapy for local control. She completed treatment after 14 months. Her end of therapy evaluation revealed that she was in remission. About 12 - 13 months after end of therapy, during routine off therapy follow-up, she was noted to be tired with leg pain (see Table 1). Her physical exam was unremarkable. Her CBC showed a white blood cell count of 4.63 K/uL, hemoglobin 9 g/dL, platelet 199 K/uL, and 4% circulating blasts.
Patient 2 underwent a bone marrow biopsy; flow cytometry on the bone marrow aspirate revealed an abnormal myeloblast population, consisting of 3% of events. The core biopsy demonstrated a hypocellular marrow for age (50 - 60% cellularity) with trilineage dyspoiesis and 10 - 15% blasts by immunohistochemical staining with CD34 and CD117 (see figure 2). The blasts were present in small clusters and singly; myeloid and erythroid maturation was left-shifted. The immature myeloid precursors were noted to have an atypical localization in the biopsy and dyspoietic changes were observed in the aspirate; specifically, the myeloid lineage exhibited nuclear to cytoplasmic dyssynchrony, hypolobated forms, and megaloblastoid changes. An immunohistochemical stain for CD61 revealed an increase in megakaryocytes, which were noted on the aspirate to include small and hypolobated forms. FISH studies and cytogenetics revealed a monosomy of chromosome 7, consistent with a therapy-related myeloid neoplasm (see Table 2).
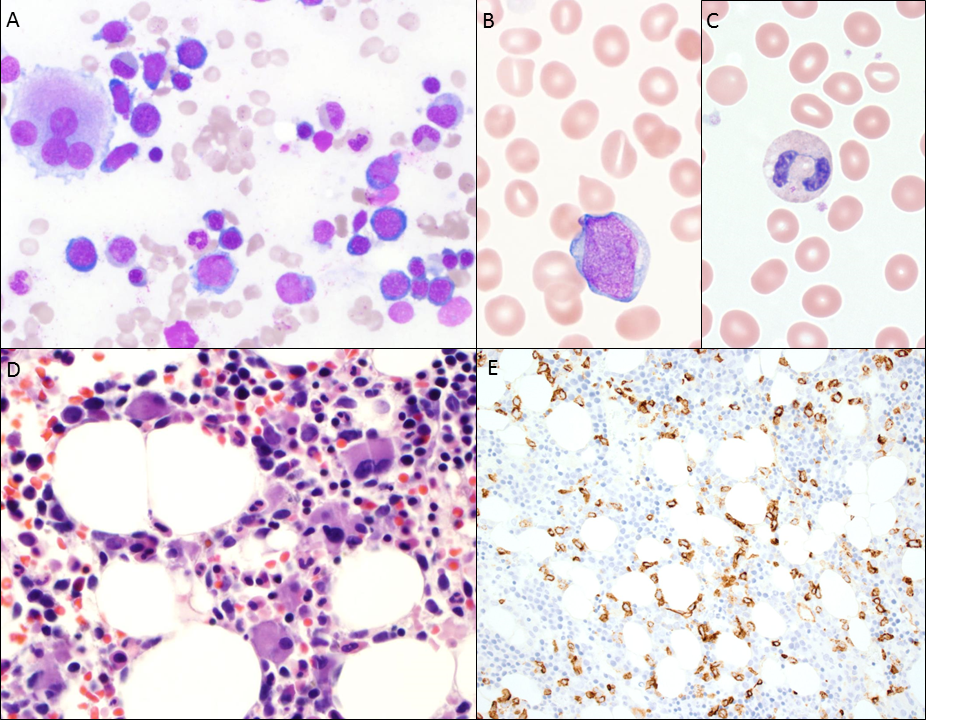
Figure 2: Patient 2 Bone Marrow Findings. A) Aspirate smear (500x) demonstrating trilineage dyspoiesis, highlighted by a megakaryocyte with widely spaced nuclei, hypogranulation of the granulocytes, and nuclear-to-cytoplasmic dyssynchrony in the erythroids. B) Peripheral smear displays scattered blasts (1000X). C) Prominent bi-lobed neutrophil on the peripheral blood smear (1000x). D) Bone marrow biopsy (400x) showing predominance of small, hypolobated, and closely distributed megakaryocytes. E) CD34 immunostain highlighting an increased blast population in the bone marrow biopsy (200x).
Patient 2 received two cycles of decitabineprior to a matched unrelated donor stem cell transplant, conditioned with fludarabine, thiotepa, rabbit ATG, and melphalan. She had no evidence of GVHD. Her peripheral blood chimerism was 100% donor by Day +25 but began to fall around Day +75, for which she received azacitidine. Unfortunately, her disease progressed to Acute Myeloid Leukemia few months later (see Figure 2), for which her family elected to place her in hospice care, and our patient expired about 3 weeks after enrolling in hospice care (see Table 1).
Discussion
Improved long-term outcomes of primary cancer have come with the cost of increased secondary malignancy[20,1]. There are some concerns that granulocyte colony stimulating factor (G-CSF) administration could play a role in leukemogenesis [21,25] of t-MNs. G-CSF is a growth factor that is central in the differentiation and amplification of neutrophil progenitors and precursors. Neutrophils are a critical part of the immune system for their antimicrobial cytotoxic properties[26]. When patients receive myelo-suppressive chemotherapy, they no longer have an adequate bone marrow reserve of myeloid precursors resulting in neutropenia. These patients are extremely susceptible to infections due to neutropenia, which can increase morbidity and mortality.
G-CSF can help in this scenario by shortening the length and decreasing the incidence of neutropenia as well as reducing the duration of antibiotic treatment[26]. Despite these impressive results, G-CSF has not shown to influence the incidence of severe infectious complications or the 5-year event free survival rate[27]. However, it has been suggested that the ability of G-CSF to promote proliferation prior to repair of damaged stem cells, which might otherwise undergo apoptosis, may contribute to leukemogenesis[21,25], facilitating growth of malignant myeloid cells and increasing the risk for t-AML and t-MDS[28].
In a European study, Navid and colleagues identified an increased cumulative incidence (5 year cumulative incidence 6.4 +/- 2.4%) of secondary leukemias (ALL and MDS/AML) in patients with ES treated with an intensified dose of alkylating agents and topoisomerase II inhibitors followed by G-CSF administration[1]. Navid et al found that most secondary leukemias occurred within 3 years from diagnosis in patients with Ewing sarcoma[1]. Galindo-Rodriguez and colleagues suggest that intensified chemotherapy used in patients with Ewing sarcoma may generate unstable clones by damaging stem cells[25]. Therapy-related myeloid neoplasm development is likely to be multifactorial and may involve several leukemogenic factors, including specific chemotherapy like topoisomerase II inhibitors and alkylators, radiation, and possibly even growth factors like G-CSF[28,11]. There is not enough long-term evidence on outcomes to make that conclusion. Prospective studies evaluating timing and dosage of G-CSF could be helpful in determining the role that G-CSF plays in the t-MN development.
Early evidence suggests that the risk of developing t-MDS/t-AML is proportional to the cumulative dose given & potentiated when different alkylators are used together or when epipodophyllotoxins or other topoisomerase II inhibiting drugs or DNA-intercalating agents (amsacrine, mitoxantrone, anthracyclines) are utilized[20]. Treatment for patients with t-MNs is challenging due to the high frequency of unfavorable cytogenetic aberrations, persistence of the primary disease, poor hematopoietic reserves, organ dysfunction, colonization with antibiotic resistant bacteria/fungi because of a chronic immunosuppressive state[16]. It is important to consider the possibility of t-MN and perform a bone marrow core biopsy with cytogenetic and molecular studies to exclude the diagnosis of rapidly evolving t-MN in patients with slow count recovery.
Conflict of Interest: There is no financial support to disclose.
References
- 1. Navid, F., Billups, C., Liu, T., et al. “Second cancers in patients with the Ewing sarcoma family of tumours.” (2008) Eur J Cancer 44(7): 983-991.
- 2. Toomey, E.C., Schiffman, J.D., Lessnick, S.L. “Recent advances in the molecular pathogenesis of Ewing’s sarcoma.” (2010) Oncogene 29(32): 4504-4516.
- 3. Laurence, V., Pierga, J.Y., Barthier, S., et al. “Long-term follow up of high-dose chemotherapy with autologous stem cell rescue in adults with Ewing tumor.” (2005) Am J Clin Oncol 28(3): 301-309.
- 4. DuBois, S.G., Grier, H.E. “Chemotherapy: the role of ifosfamide and etoposide in Ewing sarcoma.” (2009) Nat Rev Clin Oncol 6(5): 251-253.
- 5. Fuchs, B., Valenzuela, R.G., Inwards, C., et al. “Complications in long‐term survivors of Ewing sarcoma.” (2003) Cancer 98(12): 2687-2692.
- 6. Bhatia, S., Krailo, M.D., Chen, Z., et al. “Therapy-related myelodysplasia and acute myeloid leukemia after Ewing sarcoma and primitive neuroectodermal tumor of bone: A report from the Children’s Oncology Group.” (2007) Blood 109(1): 46-51.
- 7. Grier, H.E., Krailo, M.D., Tarbell, N.J., et al. “Addition of ifosfamide and etoposide to standard chemotherapy for Ewing’s sarcoma and primitive neuroectodermal tumor of bone.” (2003) New Eng J Med 348(8): 694-701.
- 8. Rodríguez-Galindo, C., Liu, T., Krasin, M.J., et al. “Analysis of prognostic factors in Ewing sarcoma family of tumors.” (2007) Cancer 110(2): 375-384.
- 9. Fuchs, B., Valenzuela, R.G., Petersen, IA., et al. “Ewing’s sarcoma and the development of secondary malignancies.” (2003) Clin Orthop Relat Res 415: 82-89.
- 10. Kuttesch, J.F. Jr, Wexler, L.H., Marcus, R.B., et al. “Second malignancies after Ewing’s sarcoma: radiation dose-dependency of secondary sarcomas.” (1996) J Clin Oncol 14(10): 2818-2825.
- 11. Rihani, R., Bazzeh, F., Faqih, N., et al. “Secondary hematopoietic malignancies in survivors of childhood cancer.” (2010) Cancer 116(18): 4385-4394.
- 12. Sultan, I., Rihani, R., Hazin, R., et al. “Second malignancies in patients with Ewing sarcoma family of tumors: a population-based study.” (2010) Acta Oncol 49(2): 237-244.
- 13. Paulussen, M., Ahrens, S., Lehnert, M., et al. “Second malignancies after Ewing tumor treatment in 690 patients from a cooperative German/Austrian/Dutch study.” (2001) Ann Oncol 12(11): 1619-1630.
PubMed||CrossRef||Others
- 14. Miser, J.S., Goldsby, R.E., Chen, Z., et al. “Treatment of metastatic Ewing sarcoma/primitive neuroectodermal tumor of bone: evaluation of increasing the dose intensity of chemotherapy—a report from the Children’s Oncology Group.” (2007) Pediatr Blood Cancer 49(7): 894-900.
- 15. Variman, J.W., Arber, D.A., Brunning, R.D., et al. Therapy-related myeloid neoplasms. In: Swerdlow SH, eds. World Health Organization Classification of Tumours of Haematopoietic and lymphoid Tissues, Revised 4th edition. Lyon, 2017:153-155.
PubMed||CrossRef||Others
- 16. Cho, H.W., Choi, Y.B., Yi, E.S., et al. “Therapy-related myeloid neoplasms in children and adolescents.” (2016) Blood Res 51(4): 242-248.
- 17. Leone, G., Fianchi, L., Pagano, L. “Incidence and susceptibility to therapy-related myeloid neoplasms.” (2010) Chem Biol Interact 184(1-2): 39-45.
- 18. Rowley, J.D., Olney, H.J. “International workshop on the relationship of prior therapy to balanced chromosome aberrations in therapy-related myelodysplastic syndromes and acute leukemia: Overview report.” (2002) Genes, Chromosomes Cancer 33(4): 331-345.
- 19. Smith, S.M., Le Beau, M.M., Huo, D., et al. “Clinical-cytogenetic associations in 306 patients with therapy-related myelodysplasia and myeloid leukemia: the University of Chicago series.” (2003) Blood 102(1): 43-52.
- 20. Barnard, D.R., Lange, B., Alonzo, T.A., et al. “Acute myeloid leukemia and myelodysplastic syndrome in children treated for cancer: comparison with primary presentation.” (2002) Blood 100(2): 427-434.
- 21. Larson, R.A. “Therapy-related myeloid neoplasms.” (2009) Haematologica 94(4): 454-459.
- 22. Pedersen-Bjergaard, J., Christiansen, D.H., Desta, F., et al. “Alternative genetic pathways and cooperating genetic abnormalities in the pathogenesis of therapy-related myelodysplasia and acute myeloid leukemia.” (2006) Leukemia 20(11): 1943-1949.
- 23. Zahid, M.F., Parnes, A., Savani, B.N., et al. “Therapy-related myeloid neoplasms-what have we learned so far?” (2016) W J Stem Cells 8(8): 231.
- 24. Kröger, N., Brand, R., van Biezen, A., et al. “Risk factors for therapy-related myelodysplastic syndrome and acute myeloid leukemia treated with allogeneic stem cell transplantation.” (2009) Haematologica 94(4): 542-549.
- 25. Rodriguez-Galindo, C., Poquette, C.A., Marina, N.M., et al. “Hematologic abnormalities and acute myeloid leukemia in children and adolescents administered intensified chemotherapy for the Ewing sarcoma family of tumors.” (2000) J Pediatr Hematol/Oncol 22(4): 321-329.
- 26. Milano-Bausset, E., Gaudart, J., Rome, A., et al. “Retrospective comparison of neutropenia in children with Ewing sarcoma treated with chemotherapy and granulocyte colony-stimulating factor (G-CSF) or pegylated G-CSF.” (2009) Clin Ther 31: 2388-2395.
- 27. Creutzig, U., Zimmermann, M., Lehrnbecher, T., et al. “Less toxicity by optimizing chemotherapy, but not by addition of granulocyte colony-stimulating factor in children and adolescents with acute myeloid leukemia: results of AML-BFM 98.” (2006) J Clin Oncol 24(27): 4499-4506.
- 28. Relling, M.V., Boyett, J.M., Blanco, J.G., et al. “Granulocyte colony-stimulating factor and the risk of secondary myeloid malignancy after etoposide treatment.” (2003) Blood 100(10): 3862-3867.










