
Development of an Automated “Just-in-Time” Derivatization Process for Gas Chromatography-Mass Spectrometry Analysis Applied to Metabolomics Samples
Colleen A. McNaney, Dieter M. Drexler*
Affiliation
Bristol-Myers Squibb Company, Research and Development, Pharmaceutical Candidate Optimization - Bioanalytical and Discovery Analytical Research, 5 Research Parkway, Wallingford, CT, USA
Corresponding Author
Dieter M. Drexler, Bristol-Myers Squibb Company, Research and Development, Pharmaceutical Candidate Optimization - Bioanalytical and Discovery Analytical Research, 5 Research Parkway, Wallingford, CT, USA, Tel: 203-677-6340; E-mail: dieter.drexler@bms.com
Citation
McNaney, C.A., et al. Development of an Automated “Just-in-Time” Derivatization Process for Gas Chromatography- Mass Spectrometry Analysis Applied to Metabolomics Samples (2016) J Anal Bioanal Sep Tech 1(1): 3- 7.
Copy rights
© 2016 Drexler, D.M. This is an Open access article distributed under the terms of Creative Commons Attribution 4.0 International License.
Keywords
Chromatography; Drug discovery; Metabolomics; Drug’s pharmacology
Abstract
The systems biology tool, metabolomics, is utilized to explore the question ”What qualitative and quantitative endogenous molecular species differentiate the samples from the respective study groups representing a control, diseased, and treated status?” utilizing a comparative analysis. Among the analytical tools, gas chromatography- mass spectrometry (GC-MS) provides in-depth data. However, the required sample preparation includes chemical derivatization steps which raise concerns about the stability of analytes, especially when dealing with large sample sets resulting in long analyses queues. To address these issues, an automated “just-in-time” derivatization methodology was developed whereby samples are processed in a “serial individual mode” resulting in high quality and reproducible data.
Introduction
Metabolomics, also referred to as metabonomics in the literature, is a systems biology tool for drug discovery and development which is being used to attain detailed knowledge of a drug’s pharmacology by obtaining mechanistic insight or discovering biomarkers which are indicative of the on-target or off-target effects of the drug under study[1,2]. Metabolomics captures the effect of an external stimulus, triggering a perturbed state, on the endogenous metabolome, which is defined as the compliment of all native small molecules (metabolites < 1,500 Da.), that participate in general metabolic reactions and that are required for the maintenance, growth and normal function of a cell. From a pharmaceutical perspective, it has been suggested that the definition also should include all small molecules introduced and modified by diet, medication, environmental exposure, and co-existing organisms. A metabolomics study (non-targeted or targeted) involves the comparative qualitative and quantitative (relative or absolute) analysis of, potentially large, sample sets from a normal state and a perturbed state, where the perturbation can be of any nature, such as genetic knockout, administration of a drug, change in diet or life style, etc.
A significant challenge is to consistently generate high quality data with excellent analytical features such as specificity, sensitivity, precision, accuracy, and reproducibility. Thus, it is important to minimize any artificially introduced variability in the samples at any step of the experiment, which is typically facilitated by following standardized and often minimalistic protocols for sample handling and preparation.
The analysis of complex samples from a metabolomics study is commonly performed using Liquid Chromatography-Mass Spectrometry (LC-MS), Nuclear Magnetic Resonance (NMR), and gas Chromatography-Mass Spectrometry (GC-MS) to obtain complementary data. While samples can be directly analyzed by LC-MS and NMR, a derivatization step is required for GC-MS analysis to improve volatility and stability of the analytes.
When employing GC-MS methodology, all samples of a study are typically manually derivatized in a “parallel batch mode” and then analyzed in a serial fashion, which raises concerns about sample stability and consequently data quality especially for large sample sets requiring overnight analyses queues with samples remaining in the auto sampler for extended periods[3,4]. To address these issues, an automated process has been developed and implemented, that performs a “serial individual mode “and “just-in-time” derivatization of the samples immediately prior to GC-MS analysis with each sample being treated in the same manner independent of its position in the sample queue thus improving data quality and confidence in the data set. Presented here are workflows and data which illustrate the benefits of this “just-in-time” sample preparation methodology as applied to the GC-MS analysis of, potentially large, sample sets from metabolomics studies.
Materials and Methods
Chemicals
Methoxylamine hydrochloride (MOX, PN226904), N-methyl-N-(trimethylsilyl) trifluoroacetamide with 1% trimethylchlorosilane (MSTFA, PN69478), pyridine (PN184527), methanol (HPLC grade), chloroform (ACS grade), isopropanol (ACS grade) were purchased from Sigma-Aldrich (St. Louis, MO).
Sample Preparation
A pool of rat plasma was split into aliquots to simulate a small metabolomics study. The samples were treated (1:2 v/v) with cold methanol (-20°C, 0.1% formic acid) to initiate a protein precipitation. The solvent also contained the internal standard (IS) to normalize for any analytical variances resultant of the sample preparation. The samples were centrifuged at 4°C for 15 minutes at 7,000 rpm. The supernatant was removed and placed into a sample vial and dried under nitrogen at 37°C until completely dry. All vials were then immediately capped with a magnetic cap with apierceable septa.
Derivatization of Samples 4
The automated process employs 2 “just-in-time” derivatizations: (1) 90 μl MOX in pyridine (40 mg/ml, step #1 derivatization reagent) was added to the dried and incubated at 80°C with gentle shaking for 15 minutes. (2) 10 μl MSTFA (step #2 derivatization reagent) was added to the sample and incubated at 80°C for 15 minutes with gentle shaking.
GC-MS Instrumentation and Software
The autosampler was a GerstelInc. (Linthicum, MD) MPS Dual Head Autosampler with ALEX (Automated Liner Exchange) option with PTV (Programmed Temperature Vaporizer) injector and was operated with Gerstel Maestro software.
An Agilent Technologies (Santa Clara, CA) 7890A gas chromatograph (GC) was interfaced with a LECO Corporation (St. Joseph, MI) Pegasus HT Time-Of-Flight MS (TOF-MS), and the GC-MS system was operated with Leco Chroma TOF software.
This setup required the parallel operation of separate sample lists in the Maestro software and the ChromaTOF software in order to implement the automated derivatization process.
The data were acquired and processed employing the ChromaTOF software. The identity of analytes in samples was confirmed with commercially available standards by comparing retention times, electron impact spectra, and entries in the NIST spectra library. For quantitative assessment, the height of the integrated peaks was utilized.
GC-MS Conditions
The GC injector was fitted with a CIS-4 baffled liner from Gerstel Inc. (PN011711-010-00). The injector conditions were as follows:1 μl injection, split mode 1:5; initial temperature at 50°C, ramp 12°C/second to 275°C and hold for 3 minutes; the PTV program was setup to substitute the used liner with a fresh liner after every 10 sample injections.
The GC column was an Rxi-5Sil MS with Integra-Guard (30 m, 0.25 mm ID, 0.50 μm with 5 m guard, PN13623-124) from Restek Corporation (Bellefonte, PA). The GC conditions were as follows: helium as carrier gas; gas flow 2.0 mL/min; initial column temperature at 100°C, hold for 6 minutes, ramp 50°C/min to 150°C and hold for 3 minutes, ramp 40°C/min to 230°C and hold for 3 minutes, ramp 40°C/min to 330°C and hold for 2.5 minutes; transfer line to MS at 280°C.
The MS conditions were as follows: electron impact (EI) mode; ion source at 250°C; acquisition delay 4 minutes; total analysis time 18 minutes; mass range 70-700 Da; acquisition rate 17 spectra/second; detector voltage was optimized with the tune function.
Results and Discussion
A metabolomics experiment seeks to answer the question “What qualitative and quantitative endogenous molecular species differentiate the respective samples from the assay defining study groups(for example, control versus diseased versus treated)?” utilizing a comparative analysis. To achieve this goal, it is imperative to minimize any potential bias or driftin the results, which might be introduced with the utilized analytical techniques.
In particular, GC-MS methodology, which requires multiple derivatizations of the analytes prior to analysis, raises concerns about sample stability, especially when dealing with the large number of samples often resultant of a metabolomics study. As depicted in schematic 1, the samples are typically derivatized in a “parallel batch mode” prior to a serial, often randomized, analysis queue; however, potentially labile and transient metabolites might be incorrectly quantified or even missed after extended scheduling intervals thus increasing the statistical data spread. To address these concerns, an automated process was developed, whereby the derivatization of the analytes is performed in a “serial individual mode” and “just-in-time” prior to analysis, so that each sample undergoes the exact same procedure including resident time of the derivatives in the autosampler - basically emulating a “single sample modeof derivatization and analysis”, which will result in improved accuracy, precision, and reproducibility of data.
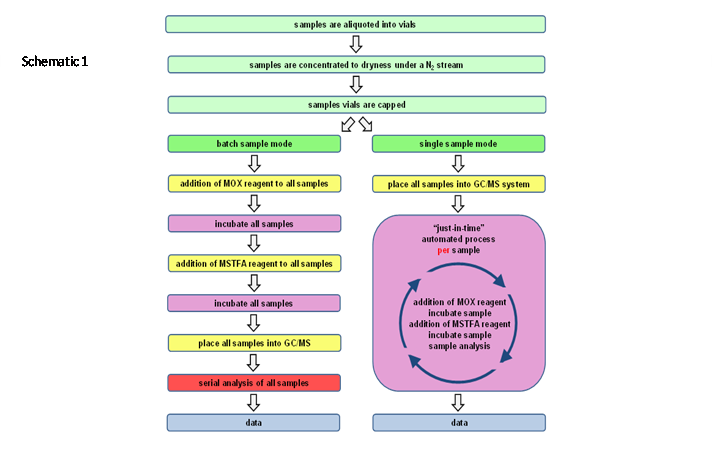
Schematic 1: Workflows for sample derivatization employing the “parallel batch mode” and the “serial individual mode” utilizing the “just-intime” approach.
The autosampler parts of the GC-MS system is displayed in Figure 1 and schematized in Figure 2. The two autosampler heads can move independently, but the control software will prevent simultaneous conflicting movements.

Figure 1: Setup of the GC-MS autosampler.
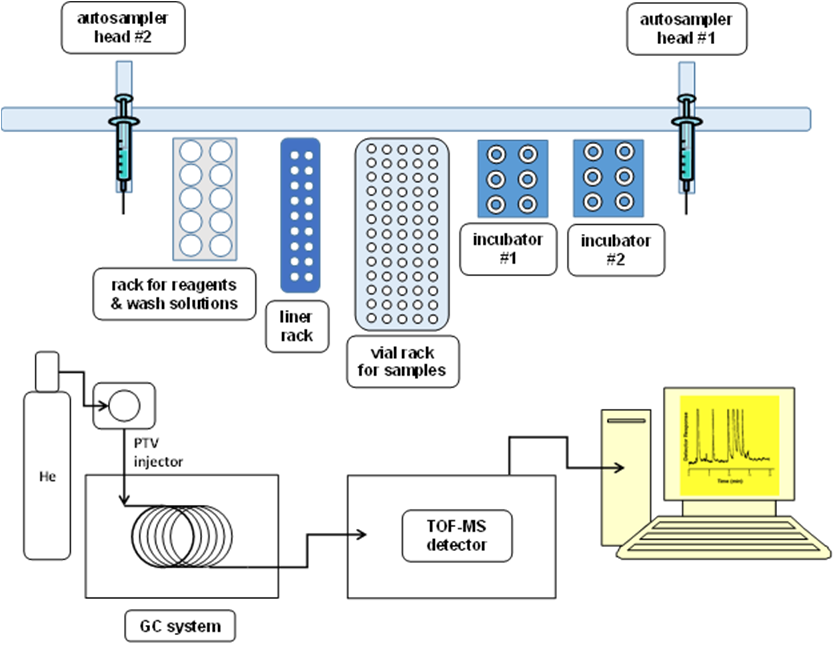
Figure 2: Layout of the GC-MS system.
The autosampler head #1 is equipped with a syringe and a magnetic tip to perform the following tasks:
• moves to the rack for reagents and aspirates step #1 derivatization reagent
• dispenses reagent into sample vial and mix
• moves sample vial to incubator #1 for a 15 minutes incubation
• moves sample vial back to vial rack
• moves to the rack for reagents and aspirates step #2 derivatization reagent
• dispenses reagent into sample vial and mix
• moves sample vial to incubator #2 for a 15 minutes incubation
• moves sample vial back to vial rack
• in-between, the syringe is washed as needed at the rack for wash solutions
The autosampler head #2 is equipped with a syringe and a liner exchange tool to perform the following tasks:
• move to the vial rack and aspirates the derivatized sample
• move to the PTV injector and inject sample to start the GC-MS analysis
• in-between, the syringe is washed as needed at the rack for wash solutions
• in-between, the GC liner is exchanged as needed
The movement of the autosampler heads and the handling of the samples is choreographed by the software to optimize the analyses of a large number of samples in the most efficient timely fashion with minimal delays. As illustrated in Figure 3, the various steps of the procedures are orchestrated in parallel workflows resulting in an automated process for “just-in-time” derivatization of the samples prior to analysis whereby each sample is identically treated thus affording high quality and reproducible data.
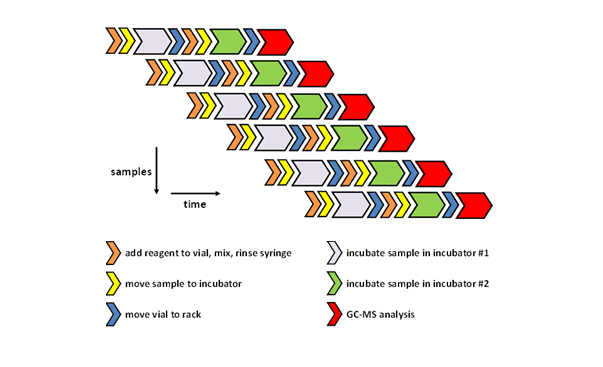
Figure 3: Parallel workflows of the “just-in-time” derivatization approach and analysis.
For a proof-of-concept study, a pool of rat plasma was split into aliquots (n = 15), simulating a small metabolomics assay, and the samples were submitted to the process (total process time ~6 hours).
In quantitative studies, an internal standard (IS) is typically employed to normalize the acquired data for analytical variance. In the case of GC-MS where a derivatization of the analyte is required, an isotopically labeled analogue of the compound will normalize for any potential issues with the reactions[5]. In this example, leucine was selected as the analyte and the 13C6-labeled analogue was used as the IS. The deuterium-labeled analogue was not suitable as the IS since the MS detection in EI mode causes the molecule to fragment into smaller subunits with the deuterium-label being stripped off, thus the analyte and the IS are not distinguishable. As shown in Figure 4, the coefficient of variation (% CV) of the analyte, as a measure of reproducibility, is exceptionally large at 47%. However, utilizing the IS computes the % CV to acceptable 12%. Expanding the IS to structural analogues of leucine (amino acids), for example, valine and alanine, the same trend is observed with excellent % CVs for the analytes.
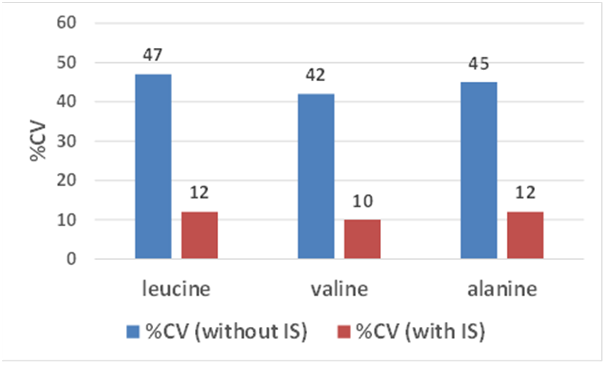
Figure 4: Reproducibility of the analyses employing an isotopically labelled analogue as internal standard.
Summary
An automated “just-in-time” derivatization process was developed for samples subjected to GC-MS analysis. This approach derivatizes and analyzes every sample in the same manner, basically a “serial individual mode”, thus addresses concerns associated with the typical manual “parallel batch mode” derivatization whereby samples might have to wait for extended periods in the autosampler prior to analysis leading to a potential degradation of analytes. This process is preferable when dealing with large sample sets, as, for example, resultant from metabolomics studies. A proof-of-concept study showed excellent reproducibility for analytes when normalized to a stable isotope labeled (SIL) analogue.
Acknowledgement:
The authors thank Drs. Petia Shipkova, SerhiyHnatyshyn, and Michael Reily for insightful discussions.
References
- 1. Drexler, D.M., Reily, M.D., Shipkova, P.A. Advances in mass spectrometry applied to pharmaceutical metabolomics. (2011) Anal Bioanal Chem 399(8): 2645-2653.
- 2. Nicholson, J.K., Wilson, I.D. Opinion: understanding 'global' systems biology: metabonomics and the continuum of metabolism. (2003) Nat Rev Drug Discov 2(8): 668-676.
- 3. Xu, F., Zou, L., Ong, C.N., et al. Experiment-originated variations, and multi-peak and multi-origination phenomena in derivatization-based GC-MS metabolomics. (2010) Trends in Anal Chem 29(3): 269-280.
- 4. Begley, P., Francis-McIntyre, S., Dunn, W. B., et al. Development and Performance of a Gas Chromatography−Time-of-Flight Mass Spectrometry Analysis for Large-Scale Nontargeted Metabolomic Studies of Human Serum. ( 2009) Anal Chem 81(16): 7038-7046.
- 5. Kaspar, H., Dettmer, K., Gronwald, W., et al. Automated GC-MS analysis of free amino acids in biological fluids. (2008) J Chromatogr B Analyt Technol Biomed Life Sci 870(2): 222-232.










