
Epidermolytic Hyperkeratosis: A Case Report from Yemen
Alshami M.A , Mohana M.J
Affiliation
- 1Dermatology Department, Faculty of Medicine and Health Sciences, University Hospital, Sana’a University, Sana’a, Yemen
- 2Faculty of Dentistry, Sana’a University, Sana’a, Yemen
Corresponding Author
Dr. Mohammad Ali Alshami, Dermatology Department, University Hospital, Sana’a University, Sana’a, Yemen, P.O. Box 1950, E-mail: mohammadalshami62@gmail.com
Citation
Alshami A.M, et al. Epidermolytic Hyperkeratosis: A Case Report from Yemen. (2016) Invest Dermatol Venerol Res 2(1): 61-63.
Copy rights
© 2016 Alshami A.M. This is an Open access article distributed under the terms of Creative Commons Attribution 4.0 International License.
Keywords
Epidermolytic hyperkeratosis; Congenital ichthyosis; Bullous congenital ichthyosiform erythroderma
Abstract
Epidermolytic hyperkeratosis is a rare autosomal dominant disorder of cornification with a prevalence of 1:100,000 to 1:300,000, which affect both sexes equally. We report the case of a 10-year-old girl who presented to the dermatology clinic with dirty brown, corrugated hyperkeratotic plaques involving joint flexures and the trunk. She had similar lesions arranged in a cobblestone pattern over the knees, elbows, and dorsal hands and feet, in addition to multiple superficial erosions. She had a history of generalized erythroderma and trauma-related blistering at the time of birth. All other family members were healthy, and had no such skin manifestations.
Introduction
Epidermolytic hyperkeratosis (EHK), also known as bullous congenital ichthyosiform erythroderma of Brocq, is a rare autosomal dominant disorder of cornification with a prevalence of 1:100,000 to 1:300,000, which affects both sexes equally, with high rates of spontaneous mutations occurring in up to 50% of the cases. The name “epidermolytic hyperkeratosis” was introduced by Frost and Van Scott for the autosomal dominant blistering form of congenital ichthyosis[1].
It presents at birth with generalized erythema, blisters, and erosions. In the months after birth, erythema and blistering decrease, while hyperkeratotic scaling that is especially prominent over the joints, neck, and dorsum of hands and feet increases. The condition is caused by mutations in either keratin 1 or keratin 10 gene, located on chromosome 12 or 17, respectively[2]. Treatment options are very limited and include urea or alpha-hydroxy acid containing creams as well as topical and systemic retinoids.
Case Report
A 10-year-old girl presented to the department of dermatology at the University Hospital in Sana’a, Yemen, with dirty brown, corrugated hyperkeratotic plaques distributed all over her body, most prominent over joint flexures, dorsal hands and feet, and neck. Similar lesions arranged in a cobblestone pattern were visible over the knees, elbows, and dorsum of hands and feet. The palms, soles, and central face were not affected. Her condition started at birth with generalized erythroderma and trauma-related blistering. By the age of 3 years, she developed dirty brown, corrugated hyperkeratotic plaques mainly over her joint flexures. Although she continued to develop erythema and blisters, such episodes decreased in severity and frequency with age. As the hyperkeratosis became more prominent, the skin developed a foul odor. Physical examination revealed thickened, brown, dirty hyperkeratotic corrugated plaques distributed over the neck, back, and flexural surfaces of her arms and legs (Figure 1- 6), affecting approximately 95% of her body surface area (Figure 1,5,6). Areas of mild erythema with minimal scaling were evident on her left lower (Figure 4,6) and upper (Figure 2) limbs. Her scalp hairs were enclosed within whitish thick scales resembling pityriasis amiantacea.
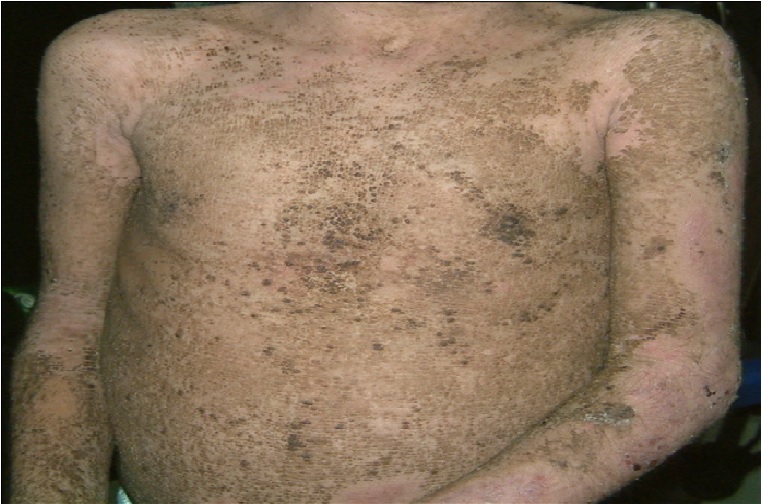
Figure 1: Dirty brown, corrugated, hyperkeratotic plaques over the trunk along with areas of superficial erosion.
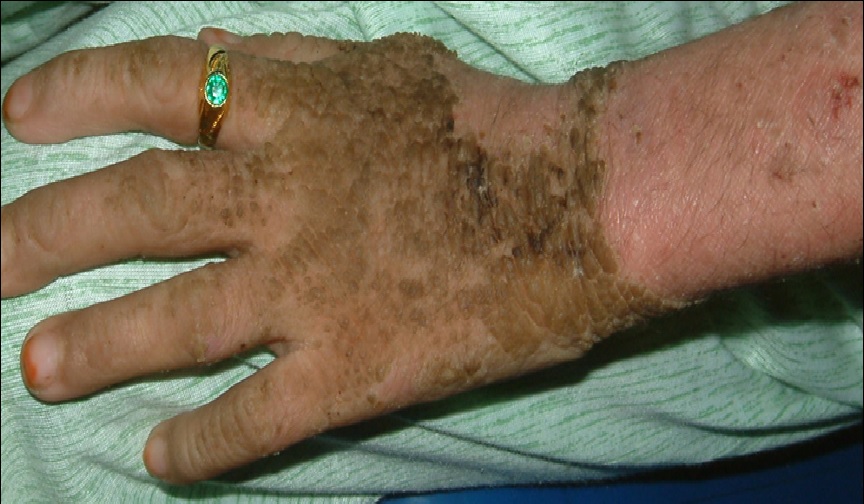
Figure 2: Dirty brown, cobblestone, hyperkeratotic plaques over the dorsum of the left hand along with areas of superficial erosion over the proximal left forearm.
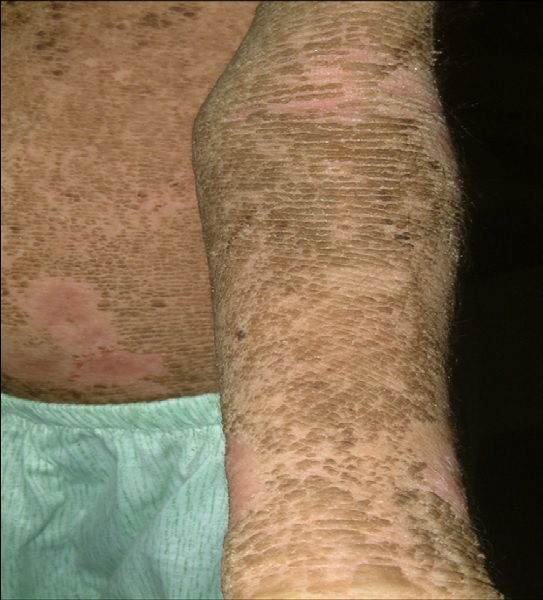
Figure 3: Prominent dirty brown, warty, corrugated hyperkeratotic plaques over the cubital fossa and volar surface of the left forearm.
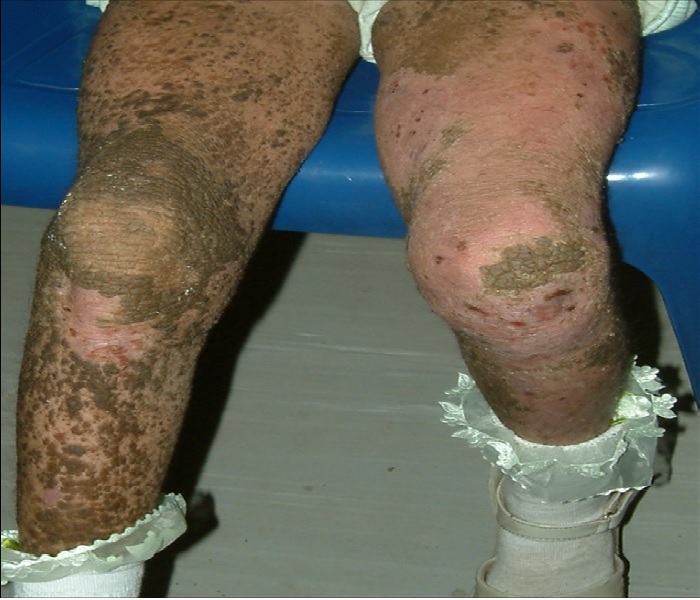
Figure 4: Hyperkeratotic warty plaques widely spread over the right lower limb, and to a lesser extent over the left lower limb, where more confluent erythema -resulting from superficial erosions is noted.
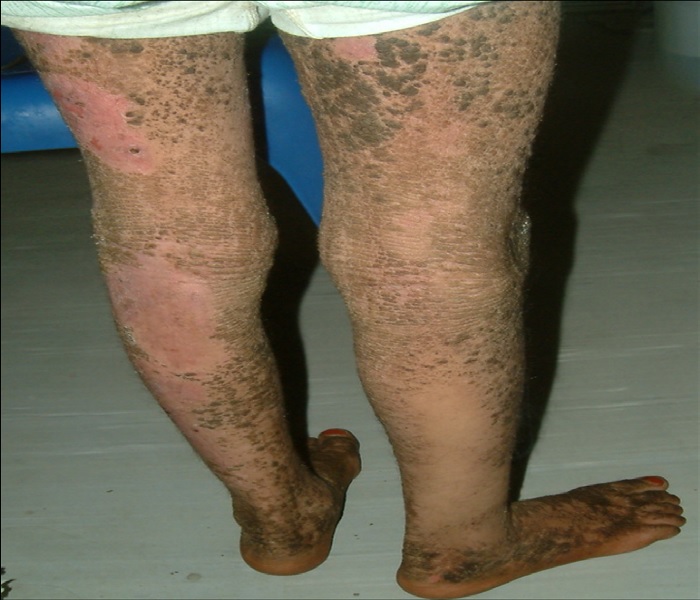
Figure 5: Dorsal aspect of the lower limbs showing large areas of dark brown hyperkeratoses alternating with areas of superficial erosion.
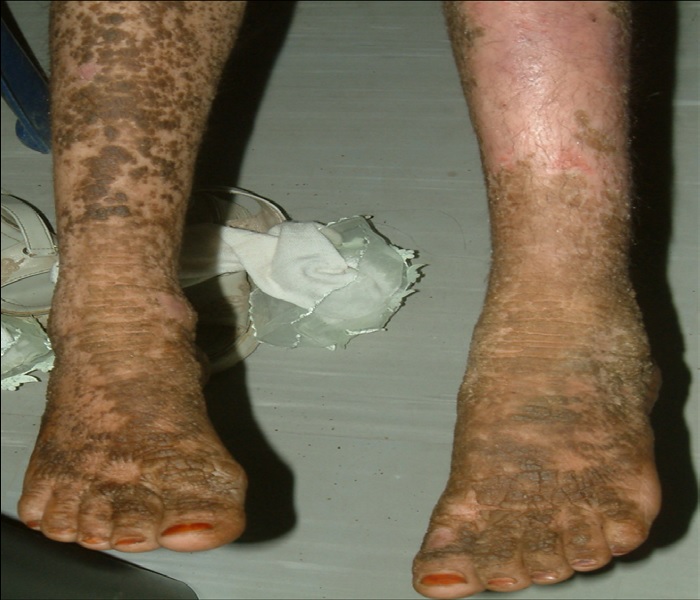
Figure 6: Anterior aspect of the legs with dirty brown, warty hyperkeratosis involving most of the right leg and dorsal feet, with the left side covered with areas of superficial erosion.
The patient was treated with 40% urea cream, which she used as tolerated. Isotretinoin could not be used in her case because of her financial background. The patient is currently being managed with topical retinoids, 40% urea cream, and topical fusidic acid.
Discussion
EHK is a rare form of congenital ichthyosis with a prevalence of 1 in 200,000 - 300,000. Brocq first described EHK in 1902, and coined the term bullous ichthyosiform erythroderma, to distinguish it from the non-blistering condition, congenital ichthyotic erythroderma[3]. EHK starts at birth with generalized erythroderma, blisters, and peeling with even mild trauma, leading to superficial ulcerations. Neonates with EHK are at risk of developing severe infections, electrolyte imbalance, and sepsis, due to disruption of the epithelial barrier. As the child grows, the erythroderma and blisters decrease, and the hyperkeratoses increase, presenting as dirty brown, verrucous, corrugated hyperkeratotic plaques more prominent on the joint flexures, but can be seen anywhere on the body. The distinct foul odor is caused by the bacterial colonization of the macerated scales, and can be treated with topical antimicrobial cleansers.
Based on the presence or absence of palmoplantar keratoderma, DiGiovanna and Bale classified EHK in 1994 into two main groups[4]. The first group involves the palms and soles (PS-type) and has three subtypes, PS1-PS3; the second group spares the palms and soles (NPS-type),and also includes three subtypes, NPS1-NPS3. Involvement of the palms and soles occurs in about 60% of patients with EHK, resulting in recurrent painful fissures and contractures that can lead to functional impairment. Our case fits into the NPS-2 type, based on the widespread involvement, the brown scales, the lack of palmoplantar keratoderma, and the lack of gait abnormality.
EHK is mostly inherited as an autosomal dominant trait, albeit 50%of cases result from spontaneous mutations, and recently an autosomal recessive inheritance has been reported[5,6]. Mutations in keratin 1 and 10 encoding genes, localized on chromosome 12 and 17, respectively, are responsible for EHK. Keratins, the major element of the cytoskeleton of keratinocytes, are intermediate filaments. Keratin mutations disrupt the keratin cytoskeleton or produce abnormal clumping of keratin filaments, leading to cytolysis of keratinocytes and blister formation with even slight trauma. Keratin 5 and 14 are expressed in the basal keratinocytes, while keratin 1 and 10 are expressed in the granular layer, which is the site of splitting in patients with EHK. Keratin 1 is co-expressed with keratin 10. Mutations in keratin 1 encoding gene are associated with severe palmoplantar keratoderma, while mutations of keratin 10 encoding gene are not[7].
A diagnosis of EHK is usually made clinically, and can be confirmed by the presence of typical histopathology. Unfortunately, a punch biopsy was refused by the parents of our patient. Immunohistochemistry and electron microscopy can be helpful in making the diagnosis, and genetic keratin studies can help in prenatal diagnosis and genetic counseling. However, these sophisticated tools are not available in most developing countries, including Yemen. It has been observed that parents with verrucous epidermal nevi, which represent somatic mosaicism for a keratin gene mutation, may have offspring with EHK[8].
The typical histopathologic features of EHK, which were first described by Nikolsky in 1897, include acanthosis, marked hyperkeratosis, coarse keratohyalin granules, and multiple perinuclear vacuoles present in the upper spinous layer[9]. Clumping of keratin intermediate filaments at the suprabasal level can be visualized by means of electron microscopy, while Immunohistochemistry can show a defect in the expression of keratin 1 and/or 10.
The pathological features are not specific for EHK, but can be seen in other benign and malignant dermatoses, such as pilar cysts, seborrheic keratosis, squamous cell carcinoma, verrucous keratosis, lichenoid amyloidosis, and granuloma annulare. Moreover, these findings can be noted in keratinizing epidermal nevi and keratoses (ichthyosis hystrix, nevus unius lateris, keratosis palmaris et plantaris, and hard nevus of Unna).
As the clinical features of EHK become more apparent with age, a wide spectrum of other genodermatoses should be considered as differentials at different stages of the disease process. In neonates, blisters and erosions can clearly differentiate EHK from non-bullous congenital ichthyosis. It can be difficult to distinguish EHK from the different variants of epidermolysis bullosa, staphylococcal scalded skin syndrome, and toxic epidermal necrolysis. Nonetheless, thorough clinical and family history, full examination, and light and electron microscopic examinations of skin biopsies as well as bacterial culture can help in making a diagnosis. During childhood, EHK can be differentiated from congenital recessive X-linked ichthyosis on the basis of the history of blistering and histological findings. Epidermolytic palmoplantar keratoderma is limited to the palms and soles, whereas ichthyosis bullosa of Siemens lacks erythroderma. Ichthyosis hystrix Curth-Macklin type patients may look like EHK patients, but there is no clinical or histological evidence of blister formation.
Management for EHK is primarily targeted to symptom control and relief. Improved neonatal care has decreased morbidity secondary to sepsis, dehydration, and electrolyte imbalances. In childhood and adulthood, malodor associated with bacterial overgrowth can be moderately controlled with antibacterial cleansers, and systemic antibiotics should be given when necessary to control infection. Topical emollients such as creams containing glycerin, lactic acid, urea, and α-hydroxy acids have been demonstrated to improve the appearance and feeling of hyperkeratotic scaly skin[10]. For more severe cases, oral and topical retinoids have also been shown to improve the skin condition, although retinoids may promote desquamation and exacerbate blistering[11]. For unknown reasons, individuals with keratin 10 gene mutations respond better to topical or systemic retinoid therapy, as compared to those with keratin 1 gene mutations.
References
- 1. Frost, P., Van Scott, E.J. Ichthyosiform dermatoses. Classification based on anatomic and biometric observations. (1966) Arch Dermatol 94(2): 113-126.
- 2. Irvine, A.D., McLean, W.H. Human keratin diseases: the increasing spectrum of disease and subtlety of the phenotype-genotype correlation. (1999) Br J Dermatol 140(5): 815-828.
- 3. Brocq, L. Erythrodermie congenitale ichthyosiforme avec hyperepidermotrophie. (1902) Ann Dermatol Syphiligre (Paris) 4:1.
- 4. DiGiovanna, J.J., Bale, S.J. Clinical heterogeneity in epidermolytic hyperkeratosis. (1994) Arch Dermatol 130(8): 1026-1035.
- 5. Terheyden, P., Grimberg, G., Hausser, I., et al. Recessive epidermolytic hyperkeratosis caused by a previously unreported termination codon mutation in the keratin 10 gene. (2009) J Invest Dermatol 129(11): 2721-2723.
- 6. Tsubota, A., Akiyama, M., Kanitakis, J., et al. Mild recessive bullous congenital ichthyosiform erythroderma due to a previously unidentified homozygous keratin 10 nonsense mutation. (2008) J Invest Dermatol 128(7): 1648-1652.
- 7. McLean, W.H., Lane, E.B. Intermediate filaments in disease. (1995) Curr Opin Cell Biol 7: 118-125.
- 8. Akhyani, M., Kiavash, K., Kamyab, K. Bullous ichthyosiform erythroderma in a child born to a parent with systematized linear epidermolytic hyperkeratosis. (2009) Int J Dermatol 48(2): 215-217.
- 9. Nikolsky, P. Contribution à l'étude des anomalies congénitales de kératinisation. (1897) Comptes-Rendus du XII Congres International de Medicine, Moscuo 4: 433-442.
- 10. Kempers, S., Katz, H.I., Wildnauer, R., et al. An evaluation of the effect of an alpha hydroxy acid-blend skin cream in the cosmetic improvement of symptoms of moderate to severe xerosis, epidermolytic hyperkatosis and ichthyosis. (1998) Cutis 61(6): 347-350.
- 11. Virtanen, M., Gedde-Dahl, T. Jr., Mork, N.J., et al. Phenotypic/genotypic correlations in patients with epidermolytic hyperkeratosis and the effects of retinoid therapy on keratin expression. (2001) Acta Derm Venereol 81(3): 163-170.










