
Following the Natural History of A Probable Adrenoleukodystrophy Case and Literature Review
Ana Carolina Andorinho de Freitas Ferreira1, André Palma Matta1, Karina Magalhães de Castro Henriques1, Osvaldo JM Nascimento1, Arielle Kirmse1, Cristina de Almeida Pereira1, Joao Dib1, Henrique Cal1, Marco Antonio Araújo Leite1, Jano Alves de Souza1, Pedro Morales1, Marco Orsini3*
Affiliation
- 1Federal Fluminense University, HUAP, Neurology Department, Niterói, Rio de Janeiro, Brazil
- 2Federal Fluminense University, HUAP, Patology Department,Niterói, Rio de Janeiro, Brazil
- 3Masters Program in Science Rehbiitation, UNISUAM, Neuroscience Department, Bonsucesso, Rio de Janeiro, Brazil
Corresponding Author
Marco Orsini, Masters Program in Science Rehabitation, UNISUAM, Neuroscience Department, Bonsucesso,Rio de Janeiro, Brazil. E-mail: orsinimarco@hotmail.com
Citation
Orsini, M., et al. Following the Natural History of a Probable Adrenoleukodystrophy( 2015) Int J Neurol Brain Disord 2(2): 1- 4.
Copy rights
© 2015 Orsini, M. This is an Open access article distributed under the terms of Creative Commons Attribution 4.0 International License.
Keywords
leukodystrophy; X-linked Adrenoleukodystrophy; Spastic paraparesis; Cognitive decline.
Abstract
Leukodystrophies compass a wide range of genetic disorders that compromise the white matter. Some of them exhibit different phenotypes with late and slow onset. The present work reports an unusual case of probable X-Linked Adrenoleukodystrophy that could be classified in adrenomyeloneupathy, but there were no signs of adrenal insufficiency and the cognitive decline developed fast. MRI evinced classical symmetrical parieto-occipital pattern of lesion, although dosage of very long chain fatty acids was normal.
Introduction
Leukodystropies (from greek words: leuko = white; dys = defective; trophy = growth) are leukoencephalopaties determined by genetic abnormalities. Since these disorders compromise white matter, magnetic resonance imaging (MRI) is of great value in showing degrees of myelin loss through grades of hypo-intensity on T1-wheighted images (lower than gray matter) and hyper-intensity in T2W images (higher than gray matter). Moreover, genetic disorders tend to let as signature a confluent, bilateral and symmetric MRI pattern[1].
Leukodystrophies are associated to a range of inborn errors of metabolism but some of them have a more indolent course. X-linked Adrenoleukody (X-ALD), a genetic disorder in which the correct oxidation of very long chain fatty acids(VLCFA) is blocked, may present with a late onset and slow progression before the patient shows severe cognitive impairment[1-3].
The present work reports a probable case of X-ALD with particularities in its natural history and diagnosis markers. The MRI was an indispensable tool to guide the investigation.
Case Report
AOB, 28 years black old man, retired for recent disability, but with normal neuropsychomotor development, started at the age of 16 with spastic paresis of his left leg that evolved to the right one. At 23 years old he was paraparetic. Two years later he started presenting sphincter dysfunction. At 28 years old, he was hospitalized with urinary infection and bradypsychism, althoughhe was still able to maintain a conversation. During the follow four months hewas in the ward, his neurological state decline severely. He kept spastic paraparesis with diffuse exaggerated deep tendon reflexes but become catatonic, responding some verbal and motor commands whit apraxia before he could just vocalize unspecific sounds when stimulated.
Exams
The first brain MRI (five years ago) only displayed discrete sulci accentuation; spinal cord MRI was normal; liquor analysis showed light protein-orrachiaand was negative for HTLV-1. Plasma serology was negative for HTLV, HIV, HBV, HCV.
Eletro-neuromyography was normal. Recent T2W MRI evinced diffuse atrophy with symmetrical hyperintensity in parieto-occipital regions including posterior limb of internal capsule and U fibers but sparing brainstem and cerebellum (Figure 1). There was no clinical or laboratorial sign of adrenal insufficiency; lipid profile was normal. Plasma concentration of VLCFA and arylsulfatase A were normal (Table 1).
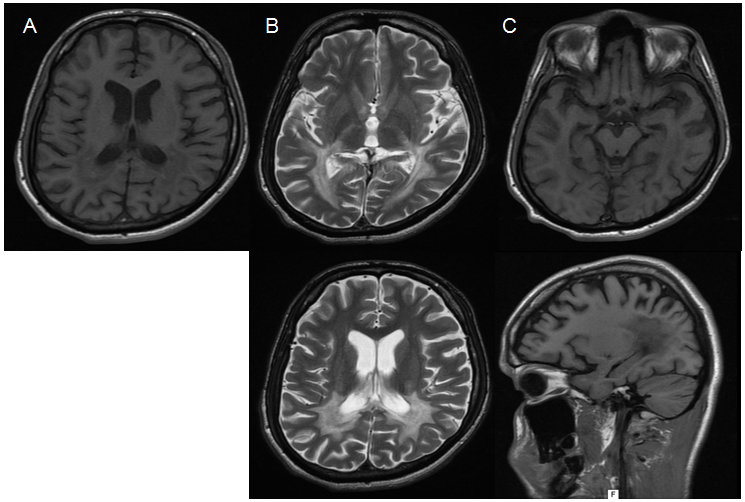
Figure 1: MRI show (A) T1W hypointensity in parieto-occipital lobes, (B) T2W hyperintensity in the same region, involving posterior limb of internal capsule (C) diffuse preeminent atrophy, extending to corpo callosum
Table 1: Biochemical analysis of plasma lipids by VLCFA method and arylsulfatase activity
| VARIABLE | ON 5TH HOSPITAL INTERNMENT | REFERENCE RANGE |
|---|---|---|
| Total Cholesterol (mg/dL) | 176 | < 200 |
| Triglycerides (mg/dL) | 41 | < 150 |
| HDL (mg/dL) | 42 | > 60 |
| LDL (mg/dL) | 132 | ≤ 130 |
| VLCFA | ||
| 24:0/22:0 | 0,92 | 0,55 – 0,89 |
| 26:0/22:0 | 0,01 | 0,004 – 0,021 |
| Hexacosanoic acid 26:0 (μmol/L) | 0,46 | 0,22 – 0,88 |
| Tetracosanoic acid 24:0 (μmol/L) | 65,8 | 44,30 – 92,40 |
| Docosanoic acid 22:0 (μmol/L) | 60,4 | 51,10 – 113,40 |
| Arylsulfatase A (nmol/h/protein mg) | 8 | 5 – 20 |
Discussion
Dysfunctions that affect predominantly the white matter of the brain are called leukoencephalopathies;when genetically determined, they are defined as eukodystrophies[1,4]. The more widely use ofMRI has been allowinginfer the etiology, while molecular genetics make specific diagnosis[4].
Although leukodystrophy is a preeminent neuropediatric field, the possible late phenotypes of some entities or the slow evolution of others make these disorders arise between all clinicians. When beginning in adulthood leukodystrophy can start with cognitive decline or behavioral changes[4,5]. The present patient initiated with motor symptoms when adolescent and just in the last couple of years he developed cognitive dysfunction. The T2W MRIshowing hyper signal in the parieto-occipital lobes symmetrically directs our investigation to adrenoleukodystrophy.
X-linked Adrenoleukodystrophyis a genetic disorder that incises in 1:17.000 newborns, preeminent in males[6,7]. The pathogenesis results from a mutation in the ABCD1 gene (located on Xq28, the terminal segment of the long arm of X chromosome) that leads to absence or abnormal expression of ALDP. This is a trans-membrane protein of peroxisome, being involved in the transport of VLCFA (> C22) to the oraganelle, where the fatty acids are beta-oxidated. If this metabolic step is impaired, VLCFA get accumulated, especially in the whitematter of the brain, the spinal cord and adrenal cortex[2,3].
In males there are the following phenotypes:
Asymptomatic status (or presymptomatic): a common and variable stage to every carrier[2,3].
A. Addison-only (Adrenocortical insufficiency): may be present in boys or men and can be the only symptom for years or even decades until neurological symptoms emerge[2,4].
B. Cerebral: is moreprevalent in childhood, which, actually, corresponds to approximately 35% of X-ALD[6]. However, this form is never registered before 2,5 years old, since it is insidious and compass visuo-spatial/ visuo-motordysfunctions or attention and reasoning impairment that may be misdiagnosed[2].Through the time, the deficits become more prominent: hyperactive behavior, apraxia, astereognosia,auditory and visual accentuated impairment, hemiparesis or spastic tetraparesis, cerebellarataxia and seizures. From this point, the outcome is fast and patient cannot do anything by himself. In adolescents and especially in adults, the evolution is slower and the psychiatric disturbances are the more evident[2].
C. Adrenomyeloneuropathy (AMN): means around 35-40% of X-ALD[6]. It represents an adult phenotype and occurs around the 3rd and 4th decade. AMNis a non-inflammatory distal axonopathyinvolves corticospinal tracts and dorsal columns of spinal cord, causing spastic paraparesis, sphincter dysfunction, sensory ataxia with impaired vibration sense. It may also affect peripheral neuropathy, causing pain[4]. AMN develops slowly (one or two decades) and manifests with lower limbs disability, sparing upper ones[2]. Adrenocortical insufficiency is concomitant in 70% of AMN patients. Brain MRI may normal, as registered in the beginning of the present case, or can display hyper signal in the pyramidal tracts along brainstem or internal capsules. According to retrospective study, only around 20% of AMN develops cerebral form in 10 years, succumbing in 1-2 years[8].
The diagnosis is complemented by MRI: Brain MRI shows hypo signal in T1 and increased signal on T2due to gadolinium enhancement in areas of demyelinating lesions. This happens in a stage when the inflammation gets severeeven if the symptoms may be absent[2,6]. In AMN, the initial lesion is a microgliosis with no significant demyelinating, hence, MRI of the spinal cord may show only non-specific atrophy and MRI may be normal. Spectroscopy may anticipate brain findings[6]. Adult cerebral phenotype usually exhibits involvement of bilateral symmetrical parieto-occipital white matter represented by hypointensity on T1WI and hyperintensity on T2WI and FLAIR images. Involvement of corpus callosum is very frequentVentricular and sulcul enlargement may be seen because of atrophy[5,9].
The reported patient could be classified in AMN form initially. He evolved with typical spastic parapaesis and urinary incontinence, but showed had no clinical or laboratorial signs of adrenal insufficiency, disturb that helps define this form. After around 10 years after the first symptom, the patient presented a rare phenotype, with very fast progression once initiated. The MRI offered classical clues to the investigation. However, the laboratory test VLCFA quantification was normal. False positive and negative is possible.It is preconized that if the level of VLCFA results increased, it should be analyzed ABCD1 mutation to confirm the diagnosis. In this case, the DNA test could be another proof but was not available.
Considering clinical manifestations and image, it is worthciting:
I. Krabbe disease: Reduced galactocerebrosidase activity, a lysosomal enzyme, leads to spastic paresis. MRI in late-onset Krabbe disease classicallyshows parieto-occipital periventricular white matter and posterior corpus callosal signal changes, but spares U fibers[2,10].
II. Metachromatic leukodystrophy: Deficiency of arylsulfatase A, another lysosomal enzyme, leads to accumulation of sulfatides in oligodendrocytes, Schwann cells and some neurons. The adult form (> 16 years old) has a slow course and mental deterioration and behavioral abnormalities are the most commonsigns[6,10]. MRI shows demyelinating periventricular, genu/splenium of the corpus callosum, descending pyramidal tracts, but spares subcortical regions and there is a frontal predominance[4,6].
III. Acyl– coenzyme A (CoA) oxidase deficiency: this is the peroxisomal enzyme that promotes the first CoA-activated-VLCFA cleavage, i.e. the β-oxidation. The enzyme has two isoforms but they are encoded by a single gene. The deficiency cause Pseudo-neonatal adrenoleukodystrophy,which presents with early craniofacial dysmorphia, generalized hypotonia, hepatomegaly, infantile seizures, loss of motor achievements, and white matter demyelination[10,11].
IV. Cerebrotendinous xanthomatosis: deficiency of 27-hydroxylase, a mitochondrial enzyme involved in bile acid synthesis, causes the increase of serum cholestanol and bile acids that deposit in the brain, lens, and tendons. The disorder manifests already in early childhood; psychomotor decline is seen later, as well as the typical tendon xanthomas[2,4].
V. Adult-Onset Autosomal Dominant Leukodystrophy (ADLD): a rare demyelinating disease of the central nervous system(CNS) caused by duplication of the lamin B1 gene (LMNB1). It is characterized by late onset (fourth or fifth decades of life) autonomic abnormalities, pyramidal and cerebellar dysfunction[12]. White matter lesions starts in the frontal lobes and extends to the cerebellum; they are symmetrical involve corticospinal tract but have a preference for brainstem with sparing of corpus callosum and periventricular region[13].
VI. Multiple sclerosis (MS): autoimmune demyelinating and degenerative disease of the CNS. Primary-progressive multiple sclerosis occurs in 15% of patients and may misdiagnosed as ADLD. However brain lesions tend to be multifocal and myelin loss has a vascular pattern, as seen inclassical lesions perpendicular to ventricular surface (Dawson’s fingers). Besides spinal cord is frequently affected[13,14].
VII. Progressive multifocal leukoencephalopathy (PML): rare myelin-degrading disease of CNS caused by thePolyomavirus, John Cunningham virus (JCV). This is an opportunistic pathogen that targets oligodendrocytes and astrocytes in immunosuppressed individuals, such as HIV/AIDS patients. The neurological impairment depends on demyelination, location, size and distribution of PML plaques. They aresometimes confluent, bilateral, and asymmetrical and may be localizedin both the gray and subcortical white matter as seen in MRI.
VIII. Human T-lymphotropic virus type 1 (HTLV-1): is responsible for myelopathy/tropical spastic paraparesis, which is defined by paraparesis, spasticity and paresthesia, lower back pain, and sphincter disturbances of insidious inception. However, specific brain lesions andcognitive deficits are uncommon[15].
The X-ALD treatment is directed particularly to mediate adrenal insufficiency. Cerebral stage progression may be arrested by allogeneic hematopoietic stem cell transplantation(HCT). It would be as anattempt to replace deficient brain microgliawith genetically competent microglial progenitor cells fromthe donor blood[2,16]. It has also been studied the autologous hematopoietic stem cells genetically correctedwith a lentiviral vector. Lorenzo’s oil acts as a competitive inhibitor of endogenous VLCFA production and has been used to postpone the cerebral form but seems to be ineffective if the symptoms are already present[2,16]. Immunomodulatory techniques have been employed with no success although VLFCA is known to induce inflammatory cytokines[6,11]. One principle for a novel therapy is the induction of the homologous ABCD2 gene encoding ABCD2 protein to compensate for ABCD1 deficiency[17]. Isotreinoin may be a future adjuvant in the up regulation of ABCD2in human monocytes/macrophages[18]. Another promise to this function would be synthetic structural analogs of thyroid hormone[16]. According to the current resources, the HCT would be the only option, but pondering the fast decline in months, it does not seem viable anymore[19].
Conclusion
The investigation of leukodystrophies requires the reunion of every available piece: from the natural history to the complementary exams, especially because occasional elements may be discordant with others and hamper specific diagnosis.
References
- 1. Schiffmann, R., van der Knaap, M.S. Invited article: an MRI-based approach to the diagnosis of white matter disorders. (2009) Neurology 72(8): 750- 759.
- 2. Engelen M., Kemp S.,de Visser M., et al., “X-linked adrenoleukodystrophy (X-ALD): clinical presentation and guidelines for diagnosis, follow-up and management.(2012) Orphanet Journal of Rare Diseases. 7(1): 51.
- 3. Shin, S.J., Kim, J.H., Kim, Y.M., et al.An Incidentally Identified Sporadic Case with Adrenoleukodystrophy with the ABCD1 Mutation.(2013) J Genet Med 10(1): 43- 46.
- 4. Leite, C.C., Lucato, L.T., Santos, G.T., et al. Imaging of adult leukodystrophies. (2014) Arq Neuropsiquiatr 72(8): 625- 632
- 5. iddiqui, S., Pawar, G., Hogg, J.P. MRI in X-linked adrenoleukodystrophy. (2015) Neurology 84(2): 211.
- 6. Eichler F., Van Haren, K. Immune response in leukodystrophies. (2007) Pediatr Neurol 37(4): 235- 244.
- 7. Engelen, M., Kemp, S. Facts on X-ALD. (2015) database X-ald.
- 8. van Geel, B.M., Bezman, L., Loes, D.J., et al. Evolution of phenotypes in adult male patients with X-linked adrenoleukodystrophy, (2001) Ann Neurol 49(2): 186– 194.
- 9. Turakhia, S., Agrawal, A. Adrenoleukodystrophy- Images.(2005) Indian J Radiol Imaging15(3): 321- 323.
- 10. Sehgal, R, Sharma, S., Sankhyan, N., et al. Teaching NeuroImages: Selective corticospinal tract involvement in late-onset Krabbe disease. (2011)Neurology77.
- 11. El Hajj, H. I., Vluggens, A., Andreoletti, P., et al. The inflammatory response in acyl-CoA oxidase 1 deficiency (pseudoneonatal adrenoleukodystrophy). (2012) Endocrinology 153(6): 2568- 2575.
- 12. Coffen C.M., McKenna C.E., Koeppen A.H, et al. Genetic localization of an autosomal dominant leukodystrophy mimicking chronic progressive multiple sclerosis to chromosome 5q31. (2000)Hum Mol Genet 9 (5): 787-793.
- 13. Beary, J., Conway, D. Adult-Onset Autosomal Dominant Leukodystrophy as a Multiple Sclerosis Mimic (P02.110). (2013)Neurology 80(Meeting Abstracts 1): P02- 110.
- 14. Hauser, S.L., Goodin. D.S. Multiple Sclerosis and other demyelinating diseases. (2012) Harrison’s principles of internal medicine 3395-3409.
- 15. Mendes, G.B., Kalil, R.S., Rosadas, C., et al. Temporal lesions and widespread involvement of white matter associated with multi-organ inflammatory disease in human T-lymphotropic virus type 1-associated myelopathy/tropical spastic paraparesis (HAM/TSP). (2014) Int J Infect Dis.
- 16. Weber, F.D., Wiesinger, C., Forss-Petter, S., et al.X-linked adrenoleukodystrophy: very long-chain fatty acid metabolism is severely impaired in monocytes but not in lymphocytes. (2014) Hum Mol Genet 23(10): 2542-2550.
- 17. Weber, F.D., Weinhofer, I., Einwich, A., et al. Evaluation of Retinoids for Induction of the Redundant Gene ABCD2 as an Alternative Treatment Option in X-Linked adrenoleukodystrophy. (2014) PLoS ONE 9(7): e103742.
- 18. Kumperscak, G.H., Paschke, E., Gradisnik, P., et al. Adult metachromatic leukodystrophy: disorganized schizophrenia-like symptoms and post-partum depression in 2 sisters. (2005)J PsychiatryNeurosci30(1): 33- 36.
- 19. Adiele, R.C., Adiele, C.A. Progressive Multifocal Leukoencephalopathy.(2014) Mult Scler.










