
Improving the Biopharmaceutical Properties of Oxcarbazepine by Co-crystallization Technique
Manoj Kumar Gautam, Divya Pandit, Yashika Bhalla, Kunal Chadha, Anupam Saini
Affiliation
University Institute of Pharmaceutical Sciences, Panjab University Chandigarh-160014, India
Corresponding Author
Renu Chadha, University Institute of Pharmaceutical Sciences, Panjab University Chandigarh-160014, India, Tel: +91-9316015096; E-mail: renukchadha@rediffmail.com
Citation
Chadha, R., et al. Improving the Biopharmaceutical Properties of Oxcarbazepine by Co-crystallization Technique. (2016) J Pharm Pharmaceutics 3(2): 96- 100.
Copy rights
© 2016 Chadha, R. This is an Open access article distributed under the terms of Creative Commons Attribution 4.0 International License.
Keywords
Co-crystallization; Oxcarbazepine; Crystal structure prediction
Abstract
The present research utilises the crystal engineering technique to modify the physiochemical properties of Oxcarbazepine (OXCBZ), antiepileptic drug used for the treatment of partial seizures with or without secondary generalization in adults and children. Crystal engineering comprises rational design and modified fabrication crystal structures and hence offers manifold prospects to selectively improve the biopharmaceutical parameters of Oxcarbazepine on the basis of indepth knowledge of crystallization processes and molecular properties. Oxcarbazepine co-crystals were evaluated for parameters by visual morphology, differential scanning calorimetry, infrared spectroscopy, X-ray diffractometry. In vitro drug release testing of co-crystals showed an increase in drug release as compared to drug release from the pure drug.
Introduction
Oxcarbazepine (OXCBZ, Trileptal [10-oxo-10, 11-dihydro-5H-dibenz (b, f)-azepine]) is a modern antiepileptic drug used as both monotherapy and adjunctive therapy for the treatment of partial seizures. Melting point of this neutral lipophilic molecule is 215 − 216°C with molecular weight 252.268 g/mol. This potential antiepileptic molecule has very low bioavailability due to its low solubility. Therefore, several formulation strategies have been tried to enhance its dissolution rate such as complexation with hydroxypropyl β-cyclodextrin[1,2], microcrystals with methylcellulose[3,4] granulation with solubility and release enhancing agents[5], solid dispersions[6]. Even few polymorphs[7,8] solvates[9] and amorphous form[10] have also been prepared to improve its solubility related problem. Pharmaceutical co-crystals are solid molecular complexes comprising an API with a neutral guest compound coformer which are a pharmaceutically acceptable or GRAS molecule within the same crystal lattice. Thus, co-crystallization is a viable means to improve the physicochemical properties such as solubility and dissolution to expand the potential of non-salt forming APIs and present manuscript is in continuous with our previous work in which two co-crystals and one solvate of oxcarbazepine[11] have been reported. The present manuscript describes the co-crystals of oxcarbazepine (OXCBZ) with nicotinic acid (NA), nicotinamide (NCT) both these counter molecules are highly soluble and are of GRAS status.
Experimental section
A gift sample was obtained from Ami Life Science Pvt. Ltd., Baroda, India. Both the coformers were of 99% purity and were procured from Hi Media Laboratories Pvt. Ltd., Mumbai, India. Solvents used were of AR grade.
Method of preparation of co-crystals
Oxcarbazepine (OXCBZ) (252 mg) was dissolved in 20 mL mixture of isopropyl alcohol and water (1:1) at 37°C. To this solution nicotinamide (NCT) (122 mg) and nicotinic acid (NA) (123 mg) were added in small portions separately. Both the solutions were stirred continuously at 37°C for 6 hours. Finally clear solutions were obtained which were lyophilized at - 20°C under reduced pressure. The dried fluffy powder was obtained and stored in desiccator under controlled condition and characterized.
The preliminary characterization was performed using Differential scanning calorimeter (DSC), Fourier-transform infrared spectroscopy (FT-IR) and Powder X-ray diffraction (PXRD).
Differential Scanning Calorimetry (DSC): DSC Q20 (TA Instruments, USA) was used for recording the thermal behavior of both the co-crystals. The samples (3 − 5 mg) were scanned at a ramping rate of 10°C/min under a dry nitrogen atmosphere (flow rate 50 mL/min). The data were collected from TA Q series Advantage software (Universal analysis 2000).
Powder X-ray Diffraction (PXRD): X’Pert PRO diffractometer system (Panalytical, Netherlands) was used to collect PXRD data. For the illumination, divergence and anti-scattering slit was set at 0.48°C. Aluminium sample holder were used to place the samples and scans were obtained between 5 and 50° in 2θ with a step size of 0.017°. X’Pert High Score software was employed for refinement of experimental PXRD pattern.
Fourier Transform-Infrared Spectroscopy (FT-IR): FTIR data was obtained from RX-I FTIR spectrometer (Perkin-Elmer, UK) in the KBr diffuse-reflectance mode (sample concentration 2 mg in 20 mg of KBr) over the range of 4000 − 400 cm−1. Data were recorded using Spectrum software.
Solid state stability studiesApproximately 200 mg of each of the co-crystals were placed in loosely capped glass vials and kept at 25°C/60% RH, 40°C/75% RH and under the ambient conditions for four weeks in humidity controlled photo stability chamber (Thermo lab, Model no. TH0000400G). Samples were then characterized by PXRD. The same procedure was repeated for the physical mixtures of OXCBZ and NCT, NA (unmilled) in a 1:1 molar ratio and monitored for any cocrystal formation by FTIR and PXRD.
Powder dissolution experiment
Powder dissolution of OXCBZ as well as OXCBZ– NA and OXCBZ–NCT was evaluated in 0.1 N HCl. To obtain an approximate particle size of 150 μm the samples were sieved using Gilson mesh sieve. A flask containing 50 mL of 0.1 N HCl was equilibrated at 37°C in a constant temperature bath in each experiment. Approximately 100 mg of the sample was added to a flask pre-equilibrated at 37°C containing 0.1 HCl. The resulting slurry was shaken at 200 rpm and aliquot at different time intervals (5, 10, 15, 20, 25, 30, 40, 50, 60, 90, 120, 150, 210, 270, and 360 min). The samples were aliquot analyzed by high performance liquid chromatography after filtering through 0.22 μm membrane filter. At the end of the experiment the pH of the slurry was measured and the remaining solids were filtered, airdried, and analyzed by PXRD.
Equilibrium solubility studies
An excess amount of drug and the co-crystals (approx. 20 mg) were shaken in 10 ml of phosphate buffer of pH 7.4 (recommended medium in USP XXV) at 37°C for 24 hours at 200 rpm in a water bath shaker (MSW-275, Macro scientific Works, Delhi). The resulting mixture with PBS was filtered and analysed by HPLC.
Crystal structure determination by PXRD
Reflex Plus module of Materials Studio (BIOVIA 7.0) was used to determine the crystal structure of the co-crystals. The various peaks in the powder diffraction pattern were indexed using X cell module and the unit cell was created and the approximate lattice parameters were depicted. It was followed by the Pawley refinement till the figure of merit reached a good quality of fit. The powder solve was performed by introducing the geometrically optimized structures (using DMol3) of the drug and coformer into the empty unit cell using a simulated annealing algorithm. It was followed by rietveld refinement and the final structure was obtained.
High performance liquid chromatography (HPLC)
The concentration of OXCBZ in pure commercial sample of OXCBZ and co-crystals (OXCBZ–NA, OXCBZ–NCT) was determined by Waters Alliance HPLC system (Waters Corporation, Milford, MA). The assembly includes a Waters 2996 Photodiode Array Detector, and a Sun Fire C18, 5 μm columns (4.6 mm × 150 mm). Pure acetonitrile was used to prepare standards of OXCBZ, NA and NCT and was diluted with 70:30 mixture of 0.1 M acetic acid/ACN to obtain various concentrations to obtain standard curve. The separation of the drug in cocrystal was performed using the mobile phase of 0.1 M acetic acid/ACN (70:30) pumped at a flow rate of 1.0 mL/min at a temperature of 35°C. The drug (OXCBZ) and the conformer (NA, NCT) were detected at 305 and 216 nm respectively[11].
Results
The prepared cocrystals of OXCBZ with nicotinamide and nicotinic acid were characterized by various techniques.
Differential scanning calorimetry (DSC)
DSC thermographs of OXCBZ showed single sharp melting endotherm at 221.98°C followed by degradation whereas NCT and NA showed a single sharp peak at 125.49°C and 238.85°C respectively (Figure 1). However, in DSC curve of OXCBZ – NCT cocrystal, a single endothermic peak appeared at 202.14°C which is in between the melting of drug and NCT. Similarly DSC curve of OXCBZ – NA cocrystal showed a single endothermic peak at 194.16°C corresponding to melting of cocrystal different from both the components. The appearance of single melting endotherm unique to both the drug and coformer suggested the formation of new solid phase in both cases. The DSC scans of physical mixture showed two individual melting peaks in both the cases corresponding to the drug and the coformer. Slight deviation from the original melting points may be due to interaction induced by thermal energy between the drug and the coformer, during the DSC scan of sample.
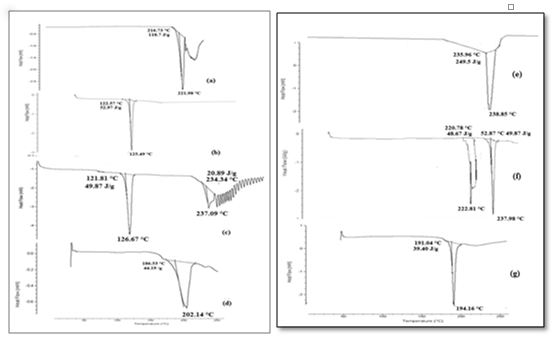
Figure 1: DSC thermograph of (a) OXCBZ (b) NCT (c) OXCBZ – NCT(PM) (d) OXCBZ – NCT (e) NA (f) OXCBZ – NA(PM) (g) OXCBZ – NA.
Fourier transform infrared spectroscopy
In FT-IR spectrum (Figure 2) of OXCBZ – NCT cocrystal, C = O stretch of carboxamide groups of OXCBZ and NCT shifted from 1652.9 cm-1 and 1680 cm-1 to 1682.7 cm-1 while N–H stretch of OXCBZ shifted from 3340 cm-1 to 3358.8 cm-1 suggesting some interaction between the amide groups of two molecules. In IR spectrum of OXCBZ – NA cocrystal, C = O stretch of amide group of OXCBZ and carboxylic group of NA at 1652.9 cm-1 and 1710 cm-1 respectively shifted to 1667.4 cm-1 while N–H stretch of OXCBZ shifted to 3336.7 cm-1 from 3340 cm-1 suggesting the formation of hydrogen bonds between –CONH and –COOH groups of OXCBZ and NA respectively. Thus, on the basis of these hydrogen bond interactions, it can be suggested that new phases formed between OXCBZ and the coformers.
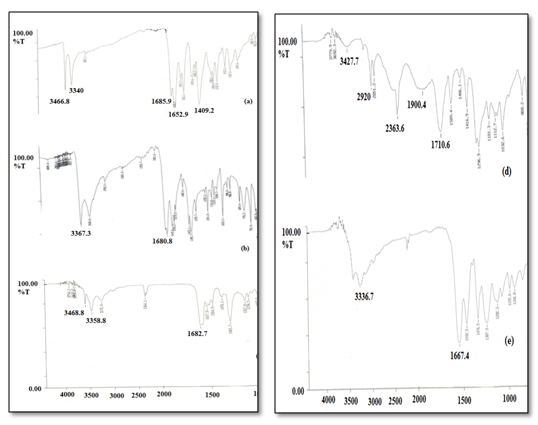
Figure 2: FT-IR spectra of (a) OXCBZ (b) NCT (c) OXCBZ – NCT (d) NA (e) OXCBZ – NA
Powder X- ray diffraction (PXRD)
OXCBZ – NCT cocrystal showed additional unique crystalline peaks (Figure 3) at 2θ values of 14.4, 14.8, 23.6, 24.9 and 26.1. Similarly, OXCBZ – NA exhibits distinguishable reflections different from those 17.6, 19.0, 20.0, 21.0, 24.8, 28.7, 32.0 and 33.0 which were absent in both the drug and the co-former. Thus presence of these additional peaks suggested the formation of new crystalline form.
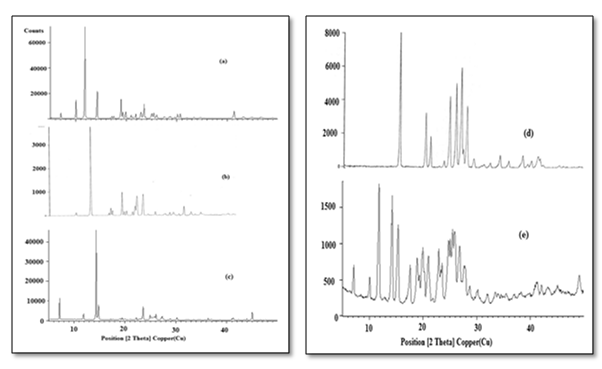
Figure 3: PXRD spectra of (a) OXCBZ (b) NCT (c) OXCBZ – NCT (d) NA (e) OXCBZ – NA
Crystal structure determination by PXRD: The crystal structure of OXCBZ–NCT was determined using PXRD, as described in the Experimental section. These co-crystals are formed as a result of heteromeric interactions between OXCBZ and coformers by replacing the homomeric interactions in OXCBZ as shown in Figure 4. OXCBZ – NCT crystallizes in the triclinic system with the space group P1. The asymmetric unit and (Figure 5), NCT is attached to OXCBZ through a hydrogen bond formed between the amide group (N5) of NCT and carbonyl group (O1’) of OXCBZ, H5N5……..O1’, However, the NCT molecule is connected with OXCBZ by hydrogen bonding between amide group at N2’ of OXCBZ and carbonyl group at O5 of NCT through H2’N2’……O5 motif. While all asymmetric units are held together by van der Waals forces. Packing pattern is given in (Figure 6a & Figure 6b).
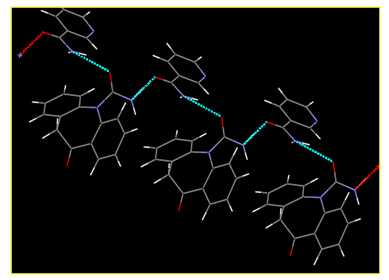
Figure 4: Hydrogen bonding interaction of OXCOZ-NCT.
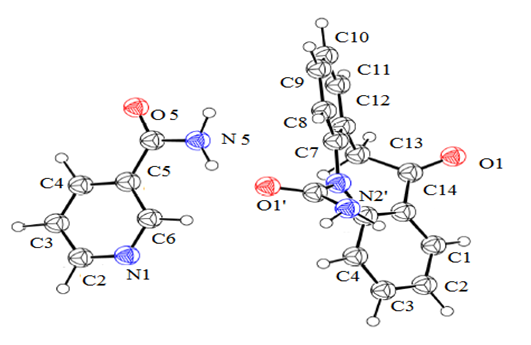
Figure 5: Asymmetric unit of OXCBZ-NCT.
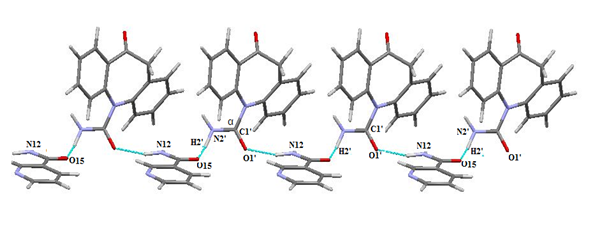
Figure 6a: Packing pattern of cocrystal OXCOZ-NCT.
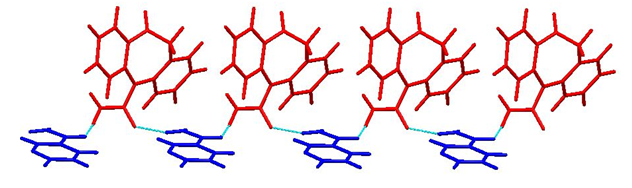
Figure 6b: Packing pattern of cocrystal OXCOZ-NCT.
Discussion
Solubility studies
The solubility of oxcarbazepine has increased from 78 mg/L to 113 mg/L in OXCBZ-NCT and 85 mg/L in OXCBZ – NA. Both the co-crystals showed improved solubility (Figure 7) in water by 1.45 folds in case of OXCBZ – NCT and 1.10 folds in case of OXCBZ – NA as compared to pure drug. Post solubility residues of both the co-crystals showed peaks of pure drugs when analysed via FTIR spectroscopy and PXRD suggesting that both the co-crystals are unstable and breaks to individual components under aqueous conditions.
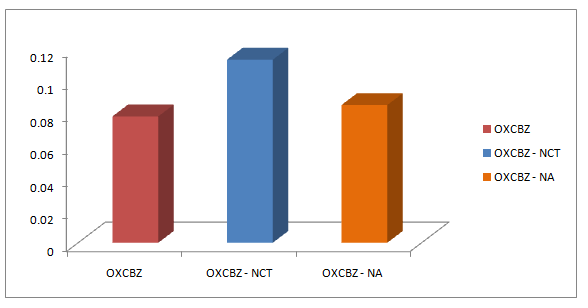
Figure 7: Solubility profile of OXCBZ and its respective cocrystals.
Intrinsic dissolution studies
Intrinsic dissolution studies were performed for the drug and co-crystals in 0.1 N HCl at 37°C for 6 hrs and concentrations determined using HPLC shows (Figure 8) the dissolution profile of co-crystals in comparison with pure drug indicating that OXCBZ – NCT exhibited maximum concentration at 30 min (0.2076 mg/mL) as compared to OXCBZ – NA (0.142 mg/mL at 15 min) and pure drug (0.132 mg/mL at 30 min). The order of dissolution profile was found to be: OXCBZ – NCT > OXCBZ – NA > OXCBZ.
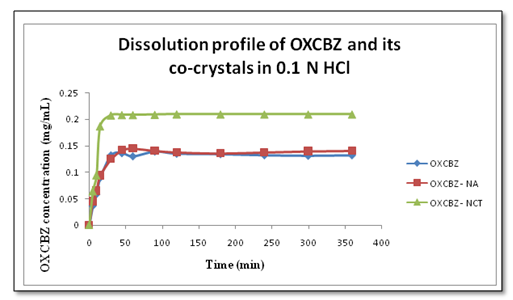
Figure 8: Dissolution profile of OXCBZ, OXCBZ – NCT and OXCBZ – NA in 0.1 N HCl at 37°C
Log P studies
The concentration of OXCBZ was measured in octanol and water at 265 nm through HPLC. The Log P of OXCBZ was 1.84; OXCBZ-NCT was 1.82 and OXCBZ-NA was1.8 respectively. Therefore, there is no significant change in log P in each solid form which suggested that formation of co-crystals do not bring any change in the lipophilicity of the drug.
Physical stability studies
Analysis of the drug and the co-crystals was performed after subjecting them to accelerated stability studies as mentioned in experimental studies. No change was observed in FTIR and PXRD of the OXCBZ-NA and OXCBZ-NCT (Figure 9). This indicates that the samples were stable even after 4 months.
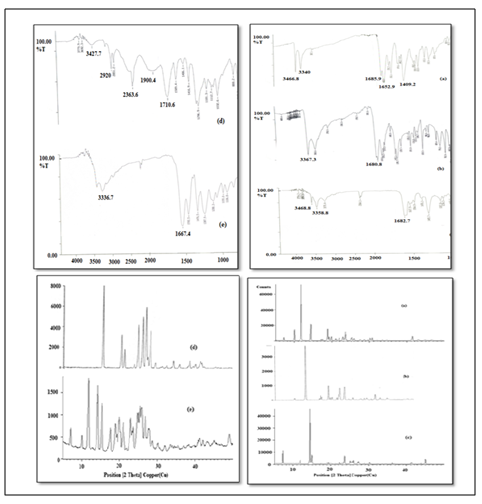
Figure 9: FIRT & PXRD data after stability studies.
In-vivo studies
In present work co-crystals of the drug were prepared with an aim to improve its solubility and hence its bioavailability. To support this hypothesis pharmacological efficacy of the drug and its co-crystals were tested by conducting MES experiment on mice after administering the dose and observing absence of hind limb extension. Results were reported as median effective dose (ED50 (mg/kg) required to protect 50% of animals tested against MES induced seizures (Figure 10). It was observed that OXCBZ – NCT and OXCBZ – NA showed ED50 of 10.8 mg/kg and 12.4 mg/kg than the drug 14.8 mg/kg suggesting that a lesser dose of co-crystals is required to prevent hind limb extension in mice in comparison to the pure drug.
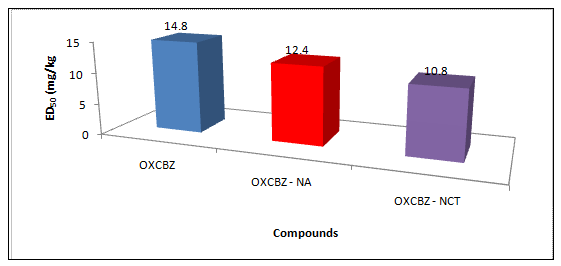
Figure 10: Comparative ED50 values of the respective newly prepared solid forms and the drug.
Conclusion
Co-crystallization has been found to be eco-friendly viable technique for improving the solubility and dissolution. Co-crystals of OXCBZ with NA and NCT were prepared by solution evaporation method and characterized by DSC, PXRD and FT-IR. The solubility of OXCBZ – NA was comparable with pure OXCBZ while it was more in case of OXCBZ – NCT (1.45 times). OXCBZ – NA was found to be unstable under ambient conditions and (40°C/75 RH) whereas OXCBZ – NCT was stable under these conditions. Based upon improved solubility of OXCBZ – NCT it was hypothesized that its absorption would also be improved due to enhanced exposure. This was confirmed by the MES tests in mice since the ED50 value for OXCBZ – NCT was lowered from 14.8 mg/kg for OXCBZ to 10.8 mg/kg.
Acknowledge:
The authors wish to thank Department of Science and Technology (DST), New Delhi (File number: SR/SI/OC-90/2012) and UGC-RGNF for providing financial support.
Conflict of interest:
None
References
- 1. Patel, N., Chotai, N., Patel, J., et al. Comparison of in-vitro dissolution profiles of oxcarbazepine-HP β-CD tablet formulations with marketed oxcarbazepine tablets. (2008) Disso Technol: 28-33.
- 2. Marziali, E., Raggi, M.A., Komarova, N., et al. Octakis‐6‐sulfato‐γ‐cyclodextrin as additive for capillary electrokinetic chromatography of dibenzoazepines: Carbamazepine, oxcarbamazepine and their metabolites. (2002) Electrophoresis 23(17): 3020-3026.
- 3. Rane, Y.M., Mashru, R.C., Sankalia, M.G., et al. Investigations on factors affecting chitosan for dissolution enhancement of oxcarbazepine by spray dried microcrystal formulation with an experimental design approach. (2007) Drug Dev Ind Pharm 33(9): 1008-1023.
- 4. Douroumis, D., Fahr, A. Enhanced dissolution of oxcarbazepine microcrystals using a static mixer process. (2007) Colloids Surf B Biointerfaces 59(2): 208-214.
- 5. Bhatt, P.P., Kidane, A., Edwards, K. Modified release preparations containing oxcarbazepine and derivatives thereof. (2011) US Patent 8017149 B2.
- 6. Douroumis, D., Bouropoulos, N., Fahr, A. Physicochemical characterization of solid dispersions of three anti-epileptic drugs prepared by solvent evaporation method. (2007) J Pharm Pharmacol 59(5): 645-653.
- 7. Aronhime, J., Dolitzky, B.Z., Berkovich, Y., et al. Crystal forms of oxcarbazepine and processes for their preparation. (2007) US Patent 7183272 B2.
- 8. Venkataraman, S., Raju, N.S., Purandhar, K., et al. Crystalline form of oxcarbazepine. (2006) US patent 0166968 A1.
- 9. Douroumis, D., Bouropoulos, N., Fahr, A. Physicochemical characterization of solid dispersions of three anti-epileptic drugs prepared by solvent evaporation method. (2007) J Pharm Pharmacol 59(5): 645-653.
- 10. Brzeczko, A.W., Doney, J.A. Amorphous oxcarbazepine and production thereof. (2008) WO Patent 092046 A2.
- 11. Chadha, R., Saini, A., Jain, D.S., et al. Preparation and Solid-State Characterization of Three Novel Multicomponent Solid Forms of Oxcarbazepine: Improvement in Solubility through Saccharin Cocrystal. (2012) Cryst Growth Des 12(8): 4211–4224.










