
In Silico Chromatography: Modeling a New Support for Alkyl-Bonded Phases and a Solvent Phase
Toshihiko Hanai
Affiliation
Health Research Foundation, Research Institute for Production Development 4F, Simogamomorimoto-cho, Sakyo-ku, Kyoto 606-0805 Japan
Corresponding Author
Toshihiko Hanai, Health Research Foundation, Research Institute for production Development 4F, Simogamomorimotocho, Sakyoku, Kyoto 606-0805, Japan; E-mail: thanai@kf7.so-net.ne.j
Citation
Hanai, T. In silico chromatography: modeling a new support for alkyl-bonded phases and a solvent phase. (2017) J Anal Bioanal Sep Tech 2(2): 111- 117.
Copy rights
© 2017 Hanai, T. This is an Open access article distributed under the terms of Creative Commons Attribution 4.0 International License.
Abstract
Chromatography is a tool to measure molecular interactions, and computational chemistry is a tool to explain molecular interactions. Therefore, reversed-phase liquid chromatography of acidic drugs using a pentyl- and an octyl-bonded silica gels was quantitatively analyzed in silico. A model pentyl- and an octyl-bonded silica gel and a methanol phases were constructed for docking with acidic drugs. Molecular interaction energy values based on van der Waals energy were obtained after docking an acidic drug into the model pentyl- or octyl-phases. Solvent effects, hydrogen bonding, and electrostatic energy values were obtained after docking an acidic drug onto the model methanol phase. Chromatographically measured log k values were correlated with the sum of van der Waals, hydrogen bonding, and electrostatic energy values. The correlation coefficient between the log k values measured using the pentyl-bonded silica gel phase and the molecular interaction energy values was 0.95 (n = 20); that between the log k values measured using the octyl-bonded silica gel and the molecular interaction energy values was 0.95 (n = 20).
Introduction
Liquid chromatography is a popular analytical technique; however, the retention mechanisms were not quantitatively explained due to the difficulty in obtaining molecular properties of the analytes. A quantitative explanation of the retention mechanisms is necessary to understand the chemistry of chromatography. The development of computational chemical calculations first permitted the prediction of log P values (octanol-water partition coefficient). The log P values were first applied to predict retention times in reversed-phase liquid chromatography, even when the molecules were ionized, if the dissociation constants (pka) were known[1]. However, log P values can only be used for the prediction of retention times in reversed-phase liquid chromatography. Therefore, further study was carried out to handle quantitative explanations of other chromatography methods. Chromatography is used to measure Molecular Interaction (MI) strengths using model compounds and is an excellent technique for measuring the relative physico-chemical values of molecules within a short time. Individual MI forces are clearly observed in different types of chromatography in terms of the different retention times of the analytes.
Chromatographic retention is described based on a combination of solubility factors using molecular properties. The selection of chromatographic mode and the column is performed based on solubility factors. The quantitative analysis of MI is of fundamental interest. Computational chemistry is a method to study molecular interactions. The development of computer software has the calculation of theoretical properties of molecules easier. Feasibility can be demonstrated using small molecules.
Computational chemical analyses provide MI energy values as the sum of mainly van der Waals, hydrogen bonding, and electrostatic energy values. The van der Waals energy is related to molecular size. The contact surface area between an analyte and an adsorbent contributes to the molecular interaction energy. Hydrogen bonding between an analyte and an adsorbent (if present) also contributes to the MI energy. When ionic interactions exist, electrostatic energy contributes to the molecular interaction energy. The measurement of direct interactions reveals the different MI strengths between an analyte and the packing material surface or the liquid phase[2-4].
In liquid chromatography, the selection of solvent is a major challenge for achieving selective separation. Even in reversed-phase liquid chromatography, the selection of solvent (organic modifier) is limited in practice; however, the selection of solvent plays a major role in chromatographic separation. In general, the computational chemical analysis of liquid chromatographic retention is performed without solvents in the calculation. Therefore, the solvation effect is obtained separately as MI between an analyte and a model solvent phase. A constructed model solvent phase is quite simple for similar compounds like derivatized amino acids[5,6] and flat molecules that do not require complete solvation for a practical approach to study the solvent effect[6,7]. However, the analysis of retention mechanisms for drugs is difficult because of their complicated stereo structure. Even a model bonded-phase was constructed, but a relation between molecular interactions and chromatographic retention was not demonstrated[8]. It seems that a construction of bonded-phase polysiloxane support was difficult; therefore, the bonded alkyl-chains were independently bent, and did not quantitatively relate with chromatographic retention. Importance of molecular interactions in chromatography was described; however, the QSRR was based on molecular properties, and they did not actually calculate molecular interactions[9].
Once model bonded-phase is constructed, the retention times of a various compounds can be quantitatively analyzed. The simplest model phase is graphitized carbon phase. It is a flat large polycyclic hydrocarbon constructed using SP2 carbon atom[10-12]. Furthermore, SP3 carbon atom can construct a honey corn type homogeneous support[13,14]. The model phase was applied to analyze retention time of phenolic compounds[15,16], drug-albumin binding affinity[17], and aromatic acid[18]. Even, model ion-exchangers were constructed and chromatographic behavior of acidic drugs on a guanidine ion exchanger[19] and basic compounds on a carboxyl ion-exchanger[20]. However, the alkyl bonded phase is dense; therefore, it supported for flat molecules such as phenolic compounds[21]. The alkyl-bonded polysiloxane support[16,22] was applied to analyze retention times in reversed-phase liquid chromatography of phenolic compounds[23], acidic drugs[24], basic drugs[25] and aromatic acids[26], and in normal-phase liquid chromatography[27].
Therefore, a simple construction method of a honey corn type support was studied. The larger atom with SP3 atomic orbital’s is lead (Pb). Even the atomic distance between two lead atoms is slightly shorter than that in the polycyclic siloxane phase. However, larger atoms having sp3 orbitals are not available in this program; therefore, lead atoms were used to build the polycyclic phase to promote in silico chromatography based on MI. Various model phases were constructed to analyze the retention mechanisms in reversed-phase liquid chromatography using pentyl- and octyl-bonded silica gel columns. The pentyl-bonded silica gel[28,29] is weak hydrophobic material and requires low concentration of organic modifier compared to popular octadecyl-bonded silica gel and used for a combination of isocratic elution and column switching technique[30].
Experimental
Chromatographic data were measured using a 100 x 2.1 mm I.D. column packed with home-made end-capped pentyl- and octyl-bonded silica gels with eluent containing 50 mM phosphoric acid and 50% methanol. The flow rate was 0.2 mL min-1. The column temperature was 40°C. Molecular modeling and molecular interaction energy values were calculated via Molecular Mechanics (MM) of CAChe™ program from Fujitsu, Japan. The computer was a PC model Prime INWIN BL672-4 with Intel Core i7 from Dospara, Yokohama, Japan. The calculated molecular properties and measured capacity ratios are summarized in [Tables I & II].
Table I: Molecular property of acidic drugs, and model pentyl- and octyl-phases.
| Compound | fs | hb | es | vw | FS1 | HB1 | ES1 | VW1 | FS2 | HB2 | ES2 | VW2 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ammoxicillinum | 55.7534 | -7.440 | 14.514 | 14.514 | -642.9581 | -12.546 | 12.884 | -957.637 | -1021.7214 | -7.751 | 15.877 | -1334.829 |
| Barbital | -59.2202 | -8.435 | -75.190 | 4.532 | -745.1419 | -9.021 | -76.310 | -948.756 | -1123.8568 | -8.535 | -75.449 | -1321.945 |
| Benzoic acid | -18.3917 | -5.701 | -6.438 | 5.269 | -707.6068 | -5.716 | -7.532 | -955.087 | -1078.6490 | -5.728 | -6.648 | -1316.648 |
| Chloamphenicol | 17.1676 | -8.459 | 25.750 | 7.412 | -677.4886 | -8.260 | 24.847 | -959.295 | -1058.1183 | -8.127 | 25.318 | -1330.480 |
| Furosemide | 0.7485 | -7.664 | -3.905 | 5.902 | -695.3108 | -7.917 | -5.191 | -959.026 | -1075.1119 | -7.378 | -4.252 | -1333.366 |
| Ibuprophen | -18.7414 | -5.805 | -4.317 | 4.763 | -708.7245 | -5.840 | -5.565 | -956.529 | -1096.4868 | -5.871 | -4.543 | -1329.776 |
| Indomethacin | -22.6674 | -5.817 | -13.530 | 6.908 | -717.8729 | -6.012 | -14.591 | -957.812 | -1099.8751 | -5.796 | -13.692 | -1331.928 |
| Iopanoic acid | -4.4765 | -4.878 | 3.836 | 6.977 | -697.7545 | -4.944 | 2.637 | -955.637 | -1081.7336 | -4.891 | 3.847 | -1332.106 |
| Mefenamic acid | 19.8971 | 0.272 | -9.420 | 19.732 | -698.1254 | -6.212 | -10.621 | -953.535 | -1083.1071 | -6.126 | -9.577 | -1325.172 |
| Nalidixiric acid | -38.4956 | -6.136 | -39.589 | 12.072 | -729.8084 | -6.145 | -40.768 | -948.636 | -1108.1258 | -6.138 | -39.409 | -1320.033 |
| Naproxen | -29.4612 | -5.811 | -4.300 | 6.878 | -727.3266 | -5.783 | -5.355 | -963.908 | -1102.5671 | -5.834 | -4.325 | -1328.668 |
| Nicotinic acid | -19.6795 | -6.029 | -9.676 | 3.879 | -707.3804 | -6.090 | -10.767 | -955.553 | -1079.0712 | -6.012 | -9.705 | -1317.546 |
| Phenylbutazone | 15.0332 | 0.000 | -10.940 | 18.105 | -680.6856 | 0.000 | -12.265 | -946.259 | -1057.9650 | 0.000 | -11.058 | -1318.433 |
| p-Hydroxylbenzoic acid | -18.5029 | -5.198 | -6.438 | 5.147 | -707.5189 | -5.329 | -7.687 | -955.100 | -1078.9363 | -5.190 | -6.383 | -1317.416 |
| Probenecid | 4.3525 | -5.704 | -3.460 | 9.257 | -704.1055 | -5.711 | -4.782 | -957.918 | -1084.7399 | -5.686 | -3.291 | -1335.265 |
| Sacylic acid | -17.4309 | -5.435 | -6.148 | 5.988 | -708.9924 | -9.278 | -7.542 | -953.755 | -1078.1121 | -5.428 | -6.402 | -1316.618 |
| Sulfamethoxazole | 0.8051 | -.028 | 2.724 | 3.083 | -696.0179 | -2.024 | 1.474 | -961.516 | -1070.6836 | -0.676 | 2.625 | -1330.728 |
| Tolazamide | -3.0540 | -2.719 | -12.840 | 8.645 | -701.1969 | -3.175 | -14.397 | -959.127 | -1089.1732 | -3.656 | -13.916 | -1337.486 |
| Tolbutamide | -29.6224 | -2.637 | -25.652 | 5.029 | -726.0762 | -2.988 | -26.999 | -963.017 | -1112.2159 | -3.847 | -27.268 | -1337.105 |
| Warfarin | -15.2297 | -0.414 | -6.057 | 7.456 | -718.0585 | -0.387 | -6.938 | -959.563 | -1095.1001 | -0.535 | -7.111 | -1330.370 |
| Pentyl-phase | -673.5968 | 0.000 | -1.237 | -945.515 | ||||||||
| Octyl-phase | -1044.2080 | 0.000 | -0.182 | -1306.013 |
fs, hb, es, vw: final structure, hydrogen bonding, electrostatic energy, van der Waals energy values (kcal mol-1); FS, HB, ES, VW: final structure, hydrogen bonding, electrostatic energy, van der Waals energy of complexes of compound and model pentyl phase (1) and octyl-phase (2).
Table II: Molecular properties of acidic drugs and model methanol phase.
| FS3 | HB3 | ES3 | VW3 | log k1 | log k2 | ||
|---|---|---|---|---|---|---|---|
| 1 | Ammoxicillinum | 15.8237 | -82.215 | -4.334 | 25.223 | -1.126 | -0.945 |
| 2 | Barbital | -103.7341 | -85.181 | -93.974 | 24.660 | -1.574 | -2.184 |
| 3 | Benzoic acid | -44.5033 | -72.340 | -21.340 | 28.528 | -0.048 | -0.471 |
| 4 | Chloamphenicol | --16.6174 | -80.336 | 8.426 | 29.895 | -0.078 | -0.049 |
| 5 | Furosemide | -34.3413 | -79.237 | -18.919 | 24.474 | -0.154 | -0.029 |
| 6 | Ibuprophen | -44.8607 | -67.032 | -20.252 | 23.871 | 1.263 | 1.429 |
| 7 | Indomethacin | --57.9789 | -73.620 | -31.658 | 26.020 | 0.882 | 1.318 |
| 8 | Iopanoic acid | -31.1429 | -68.602 | -11.130 | 26.506 | 1.404 | 1.701 |
| 9 | Mefenamic acid | -31.1087 | -66.663 | -26.136 | 34.907 | 1.404 | 1.704 |
| 10 | Nalidixiric acid | -70.7731 | -72.999 | -56.329 | 31.645 | 0.035 | 0.160 |
| 11 | Naproxen | -61.2141 | -71.250 | -20.143 | 24.754 | 0.609 | 0.793 |
| 12 | Nicotinic acid | -53.7529 | -80.020 | -26.305 | 25.319 | -1.030 | -1.013 |
| 13 | Phenylbutazone | -14.7741 | -63.096 | -28.932 | 37.119 | 0.837 | 1.155 |
| 14 | p-Hydroxylbenzoic acid | -47.9586 | -74.425 | -22.517 | 29.209 | -0.099 | -0.504 |
| 15 | Probenecid | -40.7448 | -72.894 | -19.713 | 24.291 | 0.586 | 0.721 |
| 16 | Sacylic acid | -47.7483 | -73.717 | -22.732 | 28.681 | -0.207 | -0.121 |
| 17 | Sulfamethoxazole | --33.9446 | -73.896 | -12.604 | 24.069 | -0.677 | -0.661 |
| 18 | Tolazamide | -46.4798 | -72.396 | -32.528 | 22.660 | 0.461 | 0.560 |
| 19 | Tolbutamide | -69.9539 | -71.918 | -44.747 | 18.580 | 0.407 | 0.520 |
| 20 | Warfarin | -52.9162 | -65.768 | -22.272 | 27.938 | 0.759 | 0.915 |
| Methanol-phase | -1.7160 | -46.017 | -13.695 | 26.690 |
FS, HB, ES, VW: final structure, hydrogen bonding, electrostatic energy, van der Waals energy of complexes of compound and model methanol phase (3): log k1 and log k2: log k measured using pentyl- and octyl-columns.
Results
Construction of model pentyl- and octyl-bonded phases
A large size polycyclic aromatic hydrocarbon was constructed as a model carbon phase, and the retention mechanisms of carbon columns were elucidated[2]. A silica gel model phase was used for the quantitative explanation of normal-phase liquid chromatography[2]. However, the construction of bonded-phases was not simple. Different model phases had to be constructed to improve the correlation between log k and the calculated MI energy values[2]. First, a simple, flat alkyl-phase bonded with a polycyclic hydrocarbon was constructed and used to study the retention mechanisms of phenolic compounds. Those structures are basically flat; therefore, a one-sided interaction with the flat model phase was used for the quantitative explanation of retention of phenolic compounds in reversed-phase liquid chromatography. Such simple model phase was not practical for the quantitative explanation of the retention mechanisms of acidic drugs. Therefore, a complicated alkyl-bonded silica gel phase was constructed. The stereo structure was not homogeneous; therefore, the docking of drugs required good artistic eyes and chemical institution to decide the initial position before docking[2]. The polysiloxane phase was not homogeneous. Two types of siloxane rings consisted with three SiO2 molecules. The bonded alkyl chains formed a ridge. Further study was carried out to build a homogeneous phase. Construction of a polycyclic carbon phase was simple; however, the atomic distances between the carbons that bind with alkyl-chains were too short when compared to atomic distances of the polysiloxane phase. The polycyclic silicone phase did not match the polycyclic siloxane phase.
The basic structure is the same as that of a polycyclic carbon phase. Once the basic phase was constructed, lead atoms were locked and bonded with alkyl groups. A model pentyl-bonded phase is shown in [Figure1] where benzoic acid is docked within this phase. The model phase was constructed using 419 carbon atoms, 952 hydrogen atoms, 90 oxygen atoms and 190 lead atoms including 81 pentyl and 9 methyl groups. Practically, such a dense alkyl-bonded phase cannot be synthesized. Alkyl-trichlorosilicone is used as a silyl reagent for the surface polymerization of alkyl groups, but the carbon content of the final products did not indicate poly-alkyl-bonded silica gels. It might be partly polymerized, and all silanol groups were not bonded with the silyl reagent. Alkyl-monochlorosilane has two methyl groups whose steric hindrance also interrupts the silylation of neighboring silanol groups. The existence of free silanol groups is easily observed from the chromatographic behavior of basic compounds such as pyridine[2]. Therefore, a valley shape alkyl-bonded phase was constructed by the replacement of pentyl groups with methyl groups for a practical use as a model phase of an alkyl-bonded porous silica gel.
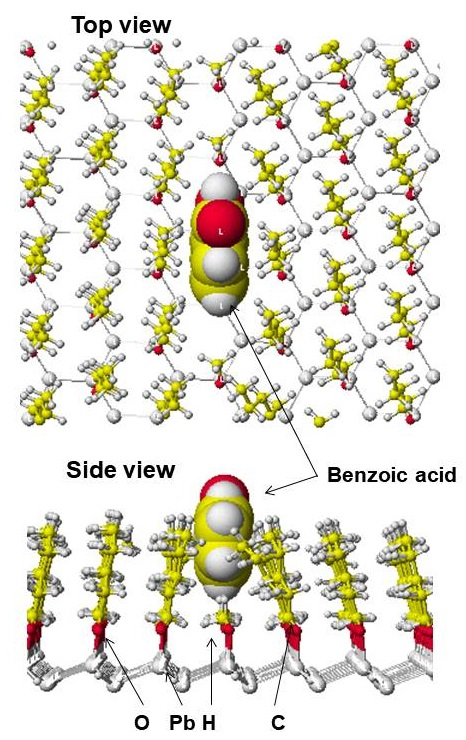
Figure 1: Figure1. Model pentyl-phase with docked benzoic acid
Atomic size of the model phase is 20% of original size.
A model octyl-bonded phase was also constructed. The first model was similar to the pentyl-bonded phase. However, this model demonstrated stronger MI with a longer molecule like phenyl butazone than small molecules and ball-type molecules. Therefore, a new model was designed that demonstrates the limited access of longer molecules and the reasonable access of ball-type molecules. The model phase is shown in [Figure 2] with benzoic acid docked inside the hole. The model phase was constructed using 473 carbon atoms, 1146 hydrogen atoms, 133 lead atoms, 60 oxygen atoms, and 63 silicone atoms including 58 octyl, 3 methyl and 2 ethyl groups.
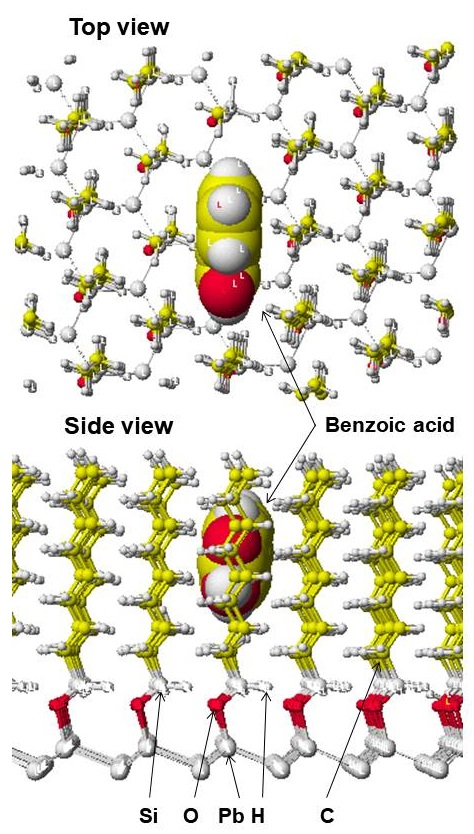
Figure 2: Model octyl-phase with docked benzoic acid
Atomic size of the model phase is 20% of original size.

Figure 3: Model methanol-phase with docked benzoic acid
Atomic size of the model phase is 20% of original size.
Construction of model methanol phases
It was difficult to construct a completely solvated phase using this computer software. A molecular dynamics program can demonstrate that a solute exists within solvent molecules; however, optimization of the structure did not produce a reproducible conformation. Therefore, a model solvent layer was constructed similar to the acetonitrile phase constructed before[2]. The flat methanol model phase did not demonstrate a good solvation model. Therefore, two flat methanol model phases were located to build a V-shape valley. The phase was constructed with 72 carbon atoms, 72 oxygen atoms and 283 hydrogen atoms. There were 72 methanol molecules in total. The carbon atoms were locked to hold the basic structure. The structure is shown in [Figure 3] with benzoic acid.
Calculation of Molecular Interaction (MI) energy values
The MI energy value (kcal mol-1) was the sum of a solute and model phase energy values minus a complex energy value. MI energy value of the final (optimized) structure (MIFS), hydrogen bonding (MIHB), electrostatic (MIES), and van der Waals (MIVW) can be calculated using the following equations.
MIFS = FS(molecule A) + FS(molecule B) – FS(molecule A and molecule B complex),
MIHB = HB(molecule A) + HB(molecule B) – HB(molecule A and molecule B complex),
MIES = ES(molecule A) + ES(molecule B) – ES(molecule A and molecule B complex),
MIVW = VW(molecule A) + VW(molecule B) – VW(molecule A and molecule B complex). The relative MIHB, MIES, and MIVW values indicate the contribution level.
The calculated Final Structure (FS), Hydrogen Bonding (HB), Electrostatic (ES), and Van Der Waals (VW) energy values of complexes with the model pentyl- and octyl phases are summarized in [Table I] with the molecular properties of the acidic drugs. MI energy values calculated using the methanol phase is given with log k values measured on both pentyl- and octyl-bonded silica gel columns in the [Table II]. Chromatography was performed in solution of pH 2; therefore, all compounds except salicylic acid were eluted in the molecular form.
MIVW calculated using the model pentyl-phase and MIHB calculated using the model methanol-phase were related with log k values measured using the pentyl-phase, and the results are given by the following equations, and shown in [Figure 4].
MIVW1 = 3.360 log k + 19.910, r = 0.611, n = 20
MIHB3 = -3.284 log k + 23.507, r = 0.827, n = 20,
MIVW1 – 2MIHB3 = 11.631 log k – 27.256, r = 0.950, n = 20.
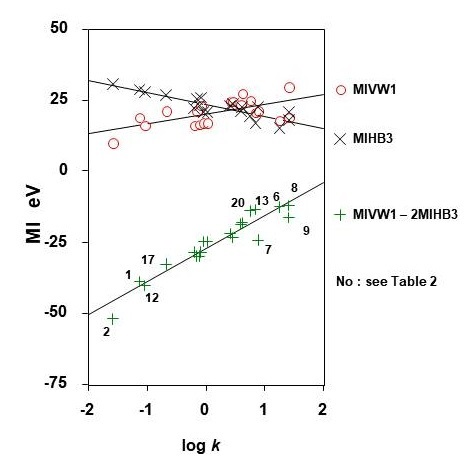
Figure 4: Correlation between molecular interaction energy values and log k values for pentyl-phase, Compound numbers: see Table 1.
The correlation coefficient for MIVW in this reversed-phase system was poor (r = 0.611, n = 20). The r values for MIHB energy in the model methanol phase was acceptable (r = 0.870, n = 20). The combined MIVW and MIHB values correlated with log k values and the r value was 0.950 (n = 20). The results indicate that the solvent effect significantly influenced reversed-phase liquid chromatography when using the pentyl-bonded silica gel column.
A similar approach was adopted for log k values measured using the octyl-bonded silica gel column. The relation between MIVW and log k and that between MIHB and log k values are given by following equations, and shown in [Figure 5].
MIVW2 = 5.150 log k + 27.996, r = 0.612, n = 20,
MIHB2 = -3.284 log k + 23.507, r = 0.827, n = 20,
MIVW2 –MIHB3 – 0.5 MIES3 = 15.119 log k – 44.128, r = 0.947, n = 20.
The contribution of the hydrophobic interaction (MIVW) is not significant (r = 0.612, n = 20), but solvation (MIHB) mainly contributed (r = 0.827, n = 20) in the reversed-phase. The combined (MIVW + MIHB) value correlated with log k values at r = 0.945 (n = 20) explained the octyl-bonded silica gel column. Addition of MIES calculated using the model methanol-phase improved the r value slightly (r = 0.947, n = 20). It seemed that MI values of indomethacin were outliers, as shown in [Figures 4 & 5]; the reason for this is not clear. The above results were recalculated. The r values improved from 0.950 to 0.964, and from 0.947 to 0.970 (n = 19) for the pentyl- and octyl-bonded phases, respectively.

Figure 5: Correlation between molecular interaction energy values and log k values for octyl-phase, Compound numbers: see Table 1.
Discussion
Designing a model phase is a critical process in silico chromatography for easy docking. A simple flat model worked well for flat molecules because one-sided MI indicated the relative MI. In general, a molecule should at least be sand-witched. A valley- type model phase was easily constructed; however, longer shaped molecules demonstrated higher MI energy than short and ball-shaped molecules, which demonstrated low MI energy values. A hole-type model was being considered as porous silica gels could be constructed; however, the hole-size was critical. A larger hole accepted a variety of compounds; however, such a hole was suitable for larger molecules but not smaller molecules. A smaller hole was not suitable for larger molecules, especially longer molecules. The octyl-phase on the above model had a moderate-sized hole; therefore a longer molecule (phenylbutazone) docked well as other molecules did, but the hole was too small for indomethacin. A large hole suitable for indomethacin was too large for smaller molecules. A valley-type model phase like the pentyl-phase was constructed; this model was suitable for phenylbutazone, but not for others. The valley-type pentyl-phase worked fine except for indomethacin. Many compounds were not covered inside the valley and parts of certain molecules were free from contact. These model phases are more homogeneous than previously proposed models[2], and the docking process was easier. However, further study of the feasibility of these model phases is required to build an auto-docking system.
Conclusion
Computational chemical analysis has been applied to the study of molecular interactions; however, some authors reported modeling without quantitatively analyzed their chromatographic data. Further, the prediction of solubility has not been achieved even in water. When the prediction of solubility is achieved, the precision of the retention time prediction will be improved using a similar computational chemical program. The above approach was a one-step improvement over previous results. As a complementary approach to these technological advances, computational chemical analysis is a promising technique with the potential to analyze the mechanisms of molecular interactions between analytes and solid phases. Especially given the feasibility of modeling the three-dimensional structures of biological macromolecules, this technology can be easily applied to study the retention mechanisms in chromatography for a variety of model phases. Additionally, theoretical calculations can provide significant insight into organic reaction mechanisms in chromatography. This method, revealing a high correlation coefficient between the measured capacity ratios and the sum of theoretically calculated molecular interaction energy and molecular property will open the possibility for the quantitative analysis of chromatographic retention mechanisms.
References
1. Hanai, T. (Ed). HPLC, A Practical Guide. (1999) Roy Soci Chemis Publish Cambrid 134.
Pubmed||Crossref||Others
2. Hanai, T. (Ed). Quantitative In Silico Chromatography, Computational Modeling of Molecular Interactions. (2014) Roy Soci Chemis Publish Cambrid 338.
Pubmed||Crossref||Others
3. Hanai, T. Introduction of in silico chromatography. (2016) J Chromatogr Sep Tech 7(5): 1- 8.
4. Hanai, T. Quantitative in silico analysis of organic modifier effect on retention in reversed-phase liquid chromatography. (2014) J Chromatogr Sci 52(1): 75-80.
5. Hanai, T. Quantitative in silico analysis of retention of phenylthiohydantoin-amino acids in reversed-phase ion-pair liquid chromatography. (2016) J Chromatogr Sci 54(4): 604-608.
6. Hanai, T., Sekine, M., Homma, H. Quantitative in silico analysis of retention of nitrobenzofurazan-amino acids in reversed-phase ion-pair liquid chromatography. (2016) J Chromatogr Sci 54(10): 1723-1726.
7. Hanai, T. in silico modeling study on molecular interactions in reversed-phase liquid chromatography. (2015) J Chromatogr Sci 53(7): 1084-1091.
8. Sander, L.C., Lippa, K.A., Wise, S.A. Order and disorder in alkyl stationary phases. (2005) Anal Bioanal Chem 382(3): 646-668.
9. Heinzen, V.E.F., Junkes, B.d.S., Kuhnen, C.A., et al. Molecular interactions in chromatographic retention: A tool for QSRR/QSPR/QSAR studies. (2012) Molecul intertact 25-48.
10. Hanai, T. Separation of polar compounds using carbon column. (2003) J Chromatogr A 989(2): 183–196.
11. Hanai, T. Analysis of mechanism of retention on graphitic carbon by a computational chemical method. (2004) J Chromatogr A 1030(1-2): 13–16.
12. Hanai, T. Quantitative in silico analysis of the specificity of graphitized (graphitic) carbons. (2011) Adv. Chromatogr 49: 257–290.
13. Hanai, T., Hatano, H., Nimura, N., et al. Computer-aided analysis of molecular recognition in chromatography. (1993) Analyst 118: 1371–1374.
14. Hanai, T., Hatano, H., Nimura, N., et al. Molecular recognition in chromatography aided by computational chemistry. (1994) Supramol Chem 3(3): 243–247.
15. Hanai, T., Koizumi, K., Kinoshita, T., et al. Prediction of pKa values of phenolic and nitrogen-containing compounds by computational chemical analysis compared to those measured by liquid Chromatography. (1997) J Chromatogr A 762(1-2): 55–61.
16: Hanai, T. Chromatography in silico, basic concept in reversed-phase liquid chromatography and computational chemistry. (2005) Anal Bioanal Chem 382(3): 708–717.
17. Hanai, T. Molecular modeling for quantitative analysis of molecular interaction. (2005) Lett Drug Des Discovery 2(3): 232–238.
18. Hanai, T. Chromatography in silico, quantitative analysis of retention mechanisms of benzoic acid derivatives. (2005) J Chromatogr A 1087(1-2): 45-51.
19. Hanai, T., Homma, H. Chromatography in silico: retention of acidic drugs on a guanidino ion-exchanger. (2007) J Liq Chromatogr Relat Technol 30(12): 1723–1731.
20. Hanai, T., Masuda Y., Homma, H. Chromatography in silico; retention of basic compounds on a carboxyl ion exchange. (2005) J Liq Chromatogr Relat Technol 28(19): 3087–3097.
21. Hanai, T. Simulation chromatography of phenolic compounds using a computational chemical method. (2004) J Chromatogr A 1027(1-2): 279-287.
22. Hanai, T. Synthesis and properties of stable bonded silica gel packings and the performance. (1996) Advances in Liquid Chromatography 1: 307–329.
23. Hanai, T., Mizutani, C., Homma, H. Computational chemical simulation of chromatographic retention of phenolic compounds. (2003) J Liq Chromatogr Relat Technol 26(13): 2031–2039.
24. Hanai, T., Miyazaki, R., Koseki, A., et al. Computational chemical analysis of the retention of acidic drugs on a pentyl-bonded silica gel in reversed-phase liquid chromatography. (2004) J Chromatogr Sci 42(7): 354–360.
25. Hanai, T. Chromatography in silico for basic drugs. (2005) J Liq Chromatogr Relat Technol 28(14): 2163–2177.
26. Hanai, T. Chromatography in silico, quantitative analysis of retention of aromatic acid derivatives. (2006) J Chromatogr Sci 44(5): 247–252.
27. Hanai, T. Quantitative in silico analysis of retention in normal-phase liquid chromatography. (2010) J Liq Chromatogr Relat Technol 33(3): 297–304.
28. Hanai, T., Dekker, M., Grushka, E., et al. Advances in Chromatography. (2000) 40: 315-357.
Pubmed||Crossref||Others
29. Ye, M., Guo, Y., Maule, B., et al. Protein/peptide separation: A comparison of stability and selectivity of C5 and C4 bonded phases, Supelco T402057.
Pubmed||Crossref||Others
30. Hanai, T., Dekker, M., Cazes Jack. Encyclopedia of Chromatography Third Edition. (2009) 480-485.
Pubmed||Crossref||Others










