
Insights into the immunopathogenesis during Japanese encephalitis virus infection
Swatantra Kumar1, Sai V. Chitti1,2 , Ravi Kant1,
Affiliation
- 1Department of Stem Cell / Cell Culture, Centre for Advance Research (CFAR), King George’s Medical University (KGMU), Lucknow 226003 (UP), India
- 2CSIR-Centre for Cellular and Molecular Biology, Uppal Road, Hyderabad 500007 (TS), India.
Corresponding Author
Prof. Shailendra K. Saxena, Deptartment of Stem Cell/Cell Culture, Centre for Advance Research (CFAR), King George’s Medical University (KGMU), Chowk, Lucknow-226003, India; Phone:+91 522 2257450 Fax: +91 522 2257450; E-mail: shailen@kgmcindia.edu
Citation
Saxena S.K., et al. Insights into the immunopathogenesis during Japanese encephalitis virus infection (2017) Cell Immunol Serum Biol 3(1): 83- 86.
Copy rights
© 2017 Saxena S.K. This is an Open access article distributed under the terms of Creative Commons Attribution 4.0 International License.
Keywords
JEV; Encephalitis; CNS infection; Immunopathogenesis; Cytokines; Chemokines
Abstract
The magnitude of Japanese encephalitis occurrence is escalating from last few years. Despite the availability of vaccine, new cases are arising; therefore it is important to look for better prevention strategies to control JEV. In order to achieve, one has to explore the various ways of immune evasion strategies employed by JEV. Persistence of JEV in the infected target cells by accumulation of autophagosome is the novel mechanism to escape host immune anti-viral response. JEV nterferes with the IFN signaling pathway by inhibiting nuclear translocation of STAT2 which is mediated by NS5 protein. JEV infection results in the up-regulation of MHC-I on the surface of infected target cells. On the other hand it also activates the cellular autophagy to diminish the innate immune response to promote the cell survival. JEV infects the central nervous system via trans-migrating neutrophils, which can breach the blood brain barrier. Detection of viral RNA via RIG-I has suggested the activation of NF-κB pathway in the infected neurons. As a result of the activation, elevated level of pro-inflammatory cytokines and chemokines contributes to the inflammation of the brain. The primary focus of this review is to discuss the immunopathogenesis during JEV infection including the future perspectives that might be crucial in understanding the involvement of various cytokines and chemokines in the development of neuroinflammation and encephalitis.
Introduction
Encephalitis is an acute inflammation of brain caused by either host immune response or viral infection. According to the World Health Organization (WHO), viral encephalitis is predominantly caused by Japanese encephalitis virus (JEV) which belongs to genus Flavivirus and family Flaviviridae. JEV is the main cause of viral encephalitis in many countries of Asia with an estimated 68,000 clinical cases every year[1,2]. JEV is a positive sense single strand (+ss) RNA virus of about 11 kb. RNA encodes a poly-protein precursor of approximately 3400 amino acid. Host cell signalases and viral encodes proteases would accomplish the processing of poly-protein and give rise to three structural proteins that includes core (C), preMembrane (prM) and envelop (E) and seven non-structural proteins NS1, NS2A, NS2B, NS3, NS4A, NS4B and NS5 which are responsible for the replication, viral transcription and also involves in alteration of host immune responses[3,4]. Interestingly, a larger NS1-related protein NS1’ (NS1 prime) is produced as a result of -1 ribosomal frame shift event, generated due to a conserved slippery heptanucleotide motif located near the upstream of NS2A gene and is induced by a downstream RNA pseudo knot structure[5].
The principle vector is Culex mosquito, most important being Culex tritaenorhynchus, present in greatest density in rainy season (June to November). Humans are accidental deadend- hosts as they do not develop a level of viraemia sufficient to infect mosquitoes. The natural cycle of JEV consists of pig-mosquito- pig or bird-mosquito-bird, and pigs serve as a biological amplifiers and reservoirs. Predominantly human JEV infection is asymptomatic or results in mild symptoms. However, a small proportion of infected individual may develop inflammation of the brain which is known as encephalitis. The manifestation of encephalitis depends on which part of the nervous system that has been affected. The early symptoms including non-specific febrile illness, diarrhea and rigor followed by symptoms such as headache, high fever, reduced level of consciousness, seizures, tremors and convulsion[6]. Many survivors of JE may acquire neuropsychiatric sequelae with cognitive and language impairment[7].
Host immune responses during JEV infection
Innate immune response against viral infection depends on rapid recognition of viral antigens by pattern recognition receptors (PRRs) which recognize pathogen associated molecular patterns (PAMPs) which are a conserved structures present on viral pathogens. Toll like receptors (TLRs) are the most well studied PRRs, which plays an important role in recognition of viral components such as nucleic acid. TLRs signaling result in the production of inflammatory cytokines, interferon (IFN) that leads to the maturation of dendritic cells and establishment of antiviral immunity. Various cell types have developed different mechanisms to sense Flavivirus infection and stimulate innate immune response. Double stranded viral RNA (dsRNA) generated during Flavivirus infection as a replication intermediate, serve as PAMPs that can be detected by PRRs. Retinoic acid-inducible gene I (RIG-I), cytoplasmic detector for dsRNA and ssRNA with 5′triphosphate. In neurons the interaction of viral RNA with RIG-I leads to the activation of various intracellular signaling pathways leading to activation of host cell cytokine gene expression and enhancement of other cellular functions such as the activation of nuclear factor κB (NF-κB)[8]. Viruses have evolved various mechanisms to disrupt the host immune system[9] such as bypass the presentation of viral antigenic peptide through MHC-I as the immune escape strategy; subsequently escape the CD8+ T lymphocyte mediated cytotoxic killing of virus infected target cell. Flaviviruses have shown to be up regulate the expression of MHC-I molecule[10] but upon JEV infection the expression of MHC-I molecule on the surface of macrophages were not significantly changed[11]. Persistence of virus, in the infected host cell, by arresting autophagy, is the novel mechanism to escape host innate immune anti-viral response. JEV activates the cellular autophagy to diminish the innate antiviral immune response and promote cell survival. However, the mechanism behind, the accumulation of autophagosome occurs during JEV replication is yet to be studied[12]. Viruses have developed mechanism to commence viral replication in the host target cells by disrupting the action of IFN and evading IFN stimulated anti-viral immune responses[13,14]. Monocytes, dendritic cells and macrophages are the most targeted cells by Flaviviruses[15]. JEV interfere with the IFN-α/β signal transduction pathway through inhibition of tyrosine phosphorylation of receptor associated kinase TYK2 and STAT2[16]. Among all non-structural proteins, NS5 is mainly responsible for the inhibition of IFN signaling pathway by interfering nuclear translocation of STAT protein (Figure 1). NS5 is the largest non-structural protein and contain most conserved, N-terminal and C-terminal domains with methyl transferase (MTase) and RNA dependent RNA polymerase (RdRp) activities respectively. IFN antagonist activity is independent of MTase and RdRp, identification of domain responsible for inhibition of IFN α/β signaling is important for determining virulence factor of JE and may be used to design potent anti-viral drugs and effective vaccine candidate[17]. Other Non-structural proteins such as NS4A and NS4B triggers membrane rearrangement during viral replication. Whereas NS2B and NS3 works together as protease which involves in deactivation of ASK1-p38MAPK signaling that would results in mitochondrial mediated apoptosis (Figure 2). Nitric oxide synthase has been shown to play an important role in protective immune response in case of viral infections. Nitric oxide (NO) prevents JEV infection by interfering with synthesis of viral RNA, accumulation of viral proteins and release of virus from infected cells. Elevated levels of inducible form of nitric oxide (iNOS) have been reported in case of JEV infection[18].
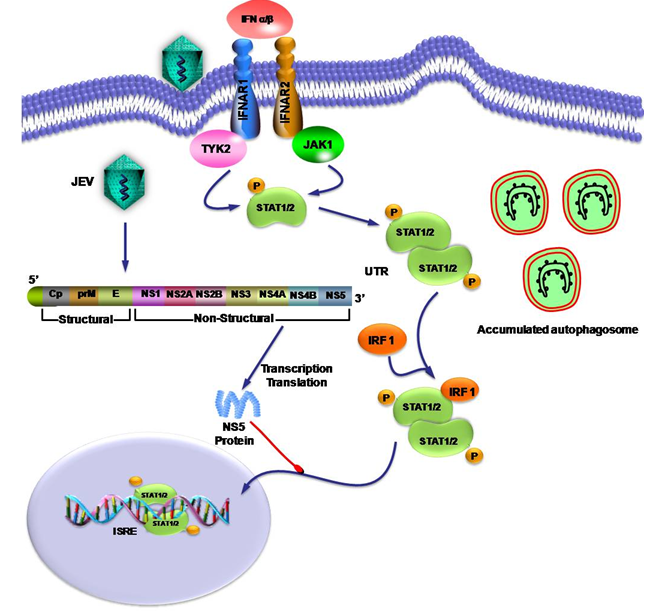
Figure 1: JEV NS5 protein mediated inhibition of inhibition of IFN-α/β signal transduction pathway.
JEV interfere with the anti-viral immune response by inhibition of IFN-α/β signal transduction pathway. NS5 protein of JEV inhibits the nuclear translocation of STAT protein from cytoplasm to nucleus and blocks interferon sensitive response elements (ISRE). JEV activates the cellular autophagy to diminish the innate antiviral immune response and promote cell survival by accumulating autophagosome.
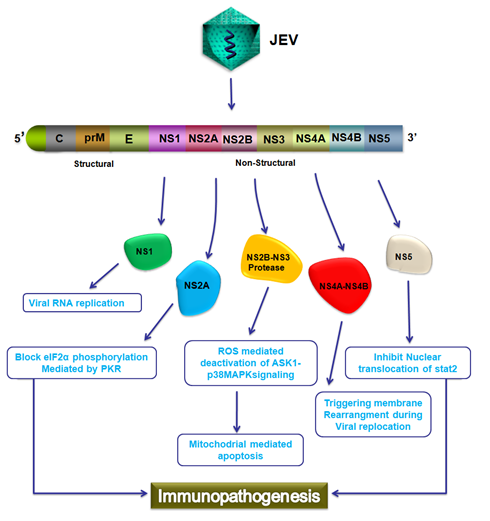
Figure 2: Schematic representation of Immunopathogenesis during JEV infection.
JEV encodes for 3400 amino acid long polyprotein which further processed and generate three structural proteins C, prM and E and seven non-structural proteins which includes NS1, NS2A, NS2B, NS3, NS4A, NS4B and NS5. NS1 protein involve in viral replication, NS2A block eIF2α phosphorylation mediated by PKR, NS2B and NS3 work as protease which involve in deactivation of ASK1 p38MAPK signaling that results in mitochondrial mediated apoptosis, NS4A and NS4B triggers membrane rearrangement during viral replication; and NS5 which involves in the IFN antagonist activity by interfering with the nuclear translocation of STAT and contribute to Immunopathogenesis.
CNS infection
Flaviviruses can cross the blood brain barrier (BBB) through passive transport mechanism across the endothelium by ‘Trojan Horse Mechanism’ in which virus is transported through the infected host inflammatory cells[19]. Central nervous system (CNS) infection is followed by leukocyte migration towards the brain matrix which often has catastrophic consequences. Chemokines have been shown to play an important role in regulating leukocytes trafficking to the sites of inflammation and tissue damage[20]. The mechanism of leukocyte infiltration is poorly understood but, it has been speculated that the pro-inflammatory cytokines and chemokines may be responsible for the cerebral recruitment and infiltration of leukocytes. Trans-migrating neutrophils can result in the production of inflammatory cytokines, reactive oxygen species (ROS), matrix metalloproteinase (MMPs) which can further degrade the basal lamina of cerebral blood vessel result in damage of BBB. Chemokines acts as a chemo-attractant for various cell types of the CNS. RANTES (encoded by CCL5 gene), is a chemokine released by CD8+ cytotoxic T lymphocytes; act as a strong chemo-attractant for T lymphocytes, monocytes, eosinophils and basophils. Viral infection has been shown to induce RANTES expression in a broad range of cells[21]. The RANTES gene promoter is divided into 5 regions (A-E) which plays distinct important role in activation of gene. The A, B and E regions of RANTES gene promoter contains binding site for transcription factors, NF-κB and NF-IL-6 respectively. Elevated levels of cytokines such as TNF-α, IL-8 and macrophage derived chemotactic factor in the serum as well as in cerebrospinal fluid has been shown upon JEV infection[22]. These elevated levels of cytokines in serum and CSF in the patients infected with JEV may be the reason behind the increased rate of mortality[23].
The activation of cell surface receptor such as C-type lectin domain family 5 member A (CLEC5A) and DAP-12 upon JEV infection result in elevated level of pro-inflammatory cytokines that causes the transmigration of leukocytes across the BBB which might be responsible for disease severity. Virus interaction with monocyte and macrophage result in strong stimulation of CLEC5A and DAP-12 mRNA expression in brain that will result in the CLEC5A/DAP-12 activation and secretion of pro-inflammatory cytokines like TNF-α and IL1-α[24]. Adhesion molecules have also been found to be over expressed during inflammation in the brain[25]. Intracellular cell adhesion molecules (ICAM) such as ICAM-1 are ligand for a receptor LFA-1 present on the leukocytes. Reports also suggest that pro-inflammatory cytokines can induce the expression of ICAM-1, leukocytes binds to the endothelial cells via ICAM-1-LFA-1 followed by the transmigration into the brain matrix[26].
Future perspectives
Emergence of Japanese encephalitis is of significant public health research concern and global awareness. The failure to overcome the spread of JE cases in Asia and Pacific region, in spite of availability of vaccine indicating us that we need to focus on effective prevention strategy. The burden of JEV infection is expected to continue to increase in the future due to climate change, globalization, socioeconomics, viral evolution and other factors. Growing cases of JEV infection in South-East Asia and other parts of the globe suggest that, there is a critical need for intense research regarding its biology and mechanism that establishes inflammation in brain and cause encephalitis. There should be expansion and strengthening of the vaccination program in the epidemic zone and areas with the highest risk of JEV transmission. JE epidemics generally occur in rural and remote settings, where diagnostic facilities are rudimentary. Currently there is no other prevention strategy other than Chemical control of vector population using insecticides shows insignificant role in JE control. There should be obligation of the environmental management for vector control for instance intermittent irrigation should be implicated, which can substantially reduce vector breeding result in reduced transmission rate. It is pertinent to execute monitoring and supervision of epidemiological areas at the field level for the successful implementation of JE. Furthermore, it is important to understand the JEV infection in CNS which may provides new insights regarding the ways to prevent neuro-inflammation and encephalitis. Since the mortality rates are mainly due to the CNS infection, if the complete entry and immunopathogenesis can be explored, one could hypothesize the major target for ideal therapeutics.
Acknowledge:
The authors are grateful to the Vice Chancellor, King George’s Medical University (KGMU), Lucknow and Director, Centre for Cellular and Molecular Biology, Council of Scientific and Industrial Research (CSIRCCMB), India for the encouragement and support for this work. SK Saxena is also supported by CCRH, Government of India, and US NIH grants: R37DA025576 and R01MH085259. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
References
- 1. Japanese Encephalitis Facts sheet. (2016). World Health Organization.
- 2. Campbell, G.L., Hills, S.L., Fischer, M., et al.estimated global incidence of Japanese encephalitis: a systematic review. (2011) Bull World Health Organ 89(10): 766-774.
- 3. Lindenbach, B.D., Rice, C.M. Molecular biology of flaviviruses. (2003) Adv Virus Res 59: 23-61.
- 4. Agrawal, P.T., Nair, M.P.N., Saxena, S.K. Japanese encephalitis: a continuing global threat. (2014) J Path Res 3(1): 042-045.
- 5. Melian, E.B., Hinzman, E., Nagasaki, T., et al. NS1' of flaviviruses in the Japanese encephalitis virus sero group is a product of ribosomal frame shifting and plays a role in viral neuroinvasiveness. (2010) J Virol 84(3): 1641-1647.
- 6. Solomon, T., Dung, N.M., Kneen, R., et al. Japanese encephalitis. (2000) J Neurol Neurosurg Psychiatry 68(4): 405-415.
- 7. Mackenzie, J.S., Gubler D.J., Petersen, L.R. Emerging flaviviruses: the spread and resurgence of Japanese encephalitis, West Nile and dengue viruses. (2004) Nat Med 10: S98-109.
- 8. Nazmi, A., Dutta, K., Basu, A. RIG-I mediates innate immune response in mouse neurons following Japanese encephalitis virus infection. (2011) PLoS One 6(6): e21761.
- 9. Saxena, S.K., Agrawal, P.T., Nair, M.P.N. Current Scenario of Antiviral Drugs for Japanese Encephalitis (2014) J Med Microb Diagn 3: 133.
- 10. Lobigs, M., Müllbacher, A., Regner, M. MHC class I up-regulation by flaviviruses: Immune interaction with unknown advantage to host or pathogen. (2003) Immunol Cell Biol 81(3): 217-223.
- 11. Kundu, K., Dutta, K., Nazmi, A., et al. Japanese encephalitis virus infection modulates the expression of suppressors of cytokine signaling (SOCS) in macrophages: Implications for the hosts’ innate immune response. (2013) Cell Immunol 285(2): 100-110.
- 12. Jin, R., Zhu, W., Cao, S., et al. Japanese encephalitis virus activates autophagy as a viral immune evasion strategy. (2013) PLoS One 8(1): e52909.
- 13. Levy, D.E., García-Sastre, A. The virus battles: IFN induction of the antiviral state and mechanisms of viral evasion. (2001) Cytokine Growth Factor Rev 12(2-3): 143-156.
- 14. García-Sastre, A. Mechanisms of inhibition of the host interferon alpha/beta-mediated antiviral responses by viruses. (2002) Microbes Infect 4(6): 647-655.
- 15. Ye, J., Zhu, B., Fu, Z.F., et al. Immune evasion strategies of flaviviruses. (2013) Vaccine 31(3): 461-471.
- 16. Lin, R.J., Liao, C.L., Lin, E., et al. Blocking of the alpha interferon-induced Jak-Stat signaling pathway by Japanese encephalitis virus infection. (2004) J Virol 78(17): 9285-9294.
- 17. Lin, R.J., Chang, B. L., Yu, H. P., et al. Blocking of interferon-induced Jak-Stat signaling by Japanese encephalitis virus NS5 through a protein tyrosine phosphatase-mediated mechanism. (2006) J virology 80(12): 5908-5918.
- 18. Saxena, S.K., Mathur, A., Srivastava, R.C. Induction of nitric oxide synthase during Japanese encephalitis virus infection: evidence of protective role. (2001) Arch Biochem Biophys 391(1): 1-7.
- 19. Diamond, M.S. Evasion of innate and adaptive immunity by flaviviruses. (2003) Immunol Cell Biol 81(3): 196-206.
- 20. Schall, T.J., Bacon, K.B. Chemokines, leukocyte trafficking, and inflammation. (1994) Curr Opin Immunol 6(6): 865-873.
- 21. Lin, Y.L., Liu, C.C., Chuang, J.I., et al. Involvement of oxidative stress, NF-IL-6, and RANTES expression in dengue-2-virus-infected human liver cells. (2000) Virology 276(1): 114-126.
- 22. Chen, C.J., Chen, J.H., Chen, S.Y., et al. Upregulation of RANTES gene expression in neuralgia by Japanese encephalitis virus infection. (2004) J Virol 78(22): 12107-12119.
- 23. Ravi, V., Parida, S., Desai, A., et al. Correlation of tumor necrosis factor levels in the serum and cerebrospinal fluid with clinical outcome in Japanese encephalitis patients. (1997) J Med Virol 51(2): 132-136.
- 24. Gupta, N., Lomash, V., Rao, P.V. Expression profile of Japanese encephalitis virus induced neuroinflammation and its implication in disease severity. (2010) J Clin Virol 49(1): 4-10.
- 25. Steffen, B.J., Breier, G., Butcher, E.C., et al. ICAM-1, VCAM-1, and MAdCAM-1 are expressed on choroid plexus epithelium but not endothelium and mediate binding of lymphocytes in vitro. (1996) Am J Pathol 148(6): 1819-1838.
- 26. Yang, L., Froio, R.M., Sciuto, T.E., et al. ICAM-1 regulates neutrophil adhesion and transcellular migration of TNF-alpha-activated vascular endothelium under flow. (2005) Blood 106(2): 584-592.










