
MRI Diagnosis in Classical Metachromatic Leukodystrophy
Rajul Rastogi*, Prabhat Kumar Bhagat, Yuktika Gupta, Shourya Sharma, Pankaj Kumar Das, Pragya Sinha, Mohini Chaudhary, Vijai Pratap
Affiliation
Department of Radiodiagnosis, Teerthanker Mahaveer Medical College and Research Center, Moradabad, U.P, India
Corresponding Author
Dr. Rajul Rastogi, MD, ADND, MNAMS, FIMSA, FICRI, Department of Radiodiagnosis, Teerthanker Mahaveer Medical College and Research Center, Moradabad, U.P, India, E-mail: eesharastogi@gmail.com
Citation
Rastogi, R., et al. MRI Diagnosis in Classical Metachromatic Leukodystrophy. (2016) Int J Neuro Brain Dis 4(1): 1- 3.
Copy rights
© 2017 Rastogi, R. This is an Open access article distributed under the terms of Creative Commons Attribution 4.0 International License.
Keywords
Metachromatic leukodystrophy; Magnetic resonance imaging; Arylsulfatase
Abstract
Metachromatic leukodystrophy (MLD) is a rare group of genetically-transmitted, autosomal-recessive, lipid-storage disorders characterised by accumulation of glycosphingolipids caused by deficiency of Arylsulfatase-A enzyme in lysosomes of cells necessary for formation of normal myelin sheath. The disease can present anytime from infancy to adulthood and is characterised by progressive deterioration of motor & neurocognitive functions with variable prognosis. Though till date, there is no definitive treatment / cure for the disease yet bone marrow transplantation in infancy has shown to delay the progression of disease & gene therapy in animal experiments has shown promising future, making early diagnosis very imperative. Diagnosis is usually suspected on magnetic resonance imaging (MRI) of brain and confirmed by enzyme assays. Hence, in this article we are describing the classical brain MRI and clinical features to make the readers aware of this uncommon but important neurological entity.
Introduction
Deficiency of Arylsulfatase-A (ARSA) enzyme in patients suffering from metachromatic leukodystrophy (MLD) results in accumulation of sulfatides within the human cells affecting their function including cells responsible for myelin-production (with predominant involvement of white matter in the brain and white matter tracts in spinal cord) and cells of biliary tract & distal renal tubules[1,2]. Microscopically, these sulfatides appears as colored granules during staining with specific dyes, hence the term metachromatic. Presence of these granules in the white matter / nerves justifies the term metachromatic leukodystrophy. This article discusses the characteristic clinico-pathological and MRI features of MLD with slight emphasis on management.
Case Report
A four-year, female child presented to pediatric outpatient department of our hospital with history of gradually progressive generalized weakness and gait disturbances since last 6 - 12 months. She recently had an episode of generalized seizure. Though there was no complaint related to speech yet it was incoherent at the time of presentation. There was no history of fever or significant perinatal history. The child was given some local treatment off & on for weakness in the village for which no medical records was available. No family history of similar symptoms / disease was noted. Routine laboratory investigations and radiograph of the chest were unremarkable.
Patient was advised MRI brain to detect the cause of her problems. It revealed near-symmetrical, bilateral, confluent, periventricular white matter, hyperintensity on T2W & T2FLAIR images at the level of centrum semiovale region with hypointense transverse striations & dots on transaxial images and corresponding hypointensity on T1W images without significant postcontrast enhancement or mass effect (Figures 1- 4). No obvious signal alteration was noted on diffusion-weighted (DW) images. No obvious sign of internal hemorrhage or calcification was noted in the white matter.
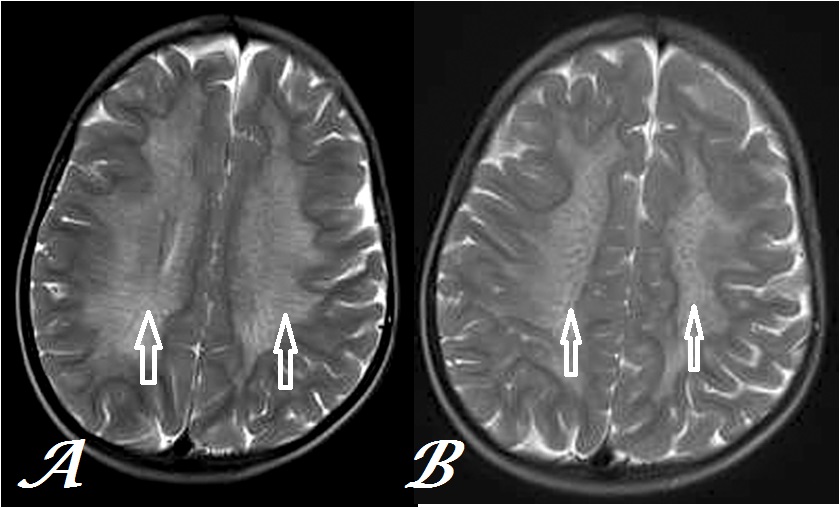
Figure 1: Transaxial, T2W MR Brain images at inferior (a) & superior (b) centrum semiovale levels show tigroid and leopard skin pattern respectively (arrows) in a case of MLD.
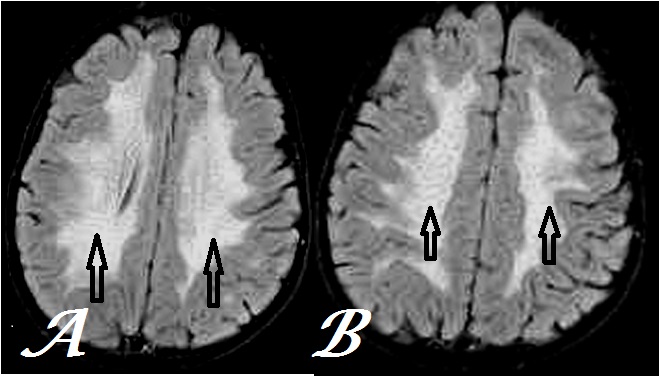
Figure 2: Transaxial, T2FLAIR MR Brain images at inferior (a) & superior (b) centrum semiovale levels show tigroid and leopard skin pattern respectively (arrows) less conspicuously than figure 1 in the same case.
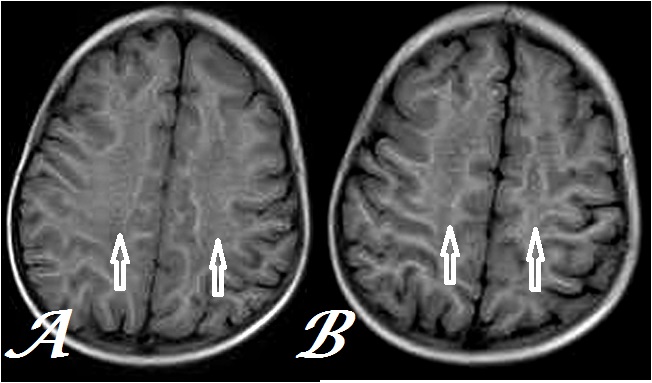
Figure 3: Transaxial, T1W MR Brain images at inferior (a) & superior (b) centrum semiovale levels show hypointensity with indistinct pattern in corresponding areas as in figure 1 (arrows) in a same case.
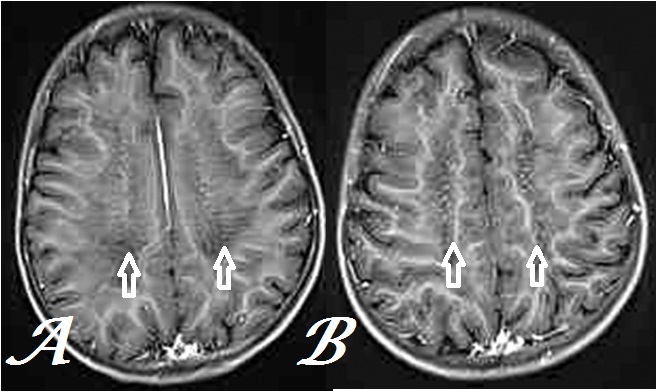
Figure 4: Transaxial, postcontrast T1GRE MR Brain images at inferior (a) & superior (b) centrum semiovale levels show conspicuous tigroid and leopard skin pattern respectively (arrows) in the same case.
Based on the above findings, the diagnosis of juvenile- form of MLD was considered and the patient blood was sent for ARSA enzyme activity in leukocytes that revealed markedly low levels (60% lower than controls), thus confirming the diagnosis. Patient was then referred to dedicated pediatric neurocentre for further management.
Discussion
MLD is known by multiple names as arylsulfatase deficiency, Greenfields’ disease, cerebroside sulfatase deficiency disease, sulfatide lipidosis, sulfatidosis, etc. MLD has a reported occurrence of up to one in 1.7 lakh worldwide with higher incidence in small population subgroup of Israel (Jews & Arabs) and in Navajo Nation[3]. MLD occur secondary to mutation in ARSA gene in lysosomes leading to deficiency of ARSA enzyme with consequent accumulation of sulfatides within the cells[4]. This gene is transmitted in autosomal recessive pattern with carriers being asymptomatic. Some of these patients show mutation in PRAP gene which results in cellular deficiency of protein, Saposin-B that works in conjunction with ARSA enzyme. Some individuals with low-ARSA levels (less than 15% of control / normal) do not reveal signs of MLD, hence referred to as pseudoarylsulfatase deficiency[5]. Accumulation of sulfatides in myelin-producing cells results in abnormal myelin-sheath in patients of MLD affecting white matter of brain as well as tracts & nerves of brain & spinal cord, involving cranial as well as peripheral nerves. Hence, these patients often present with pseudobulbar palsies, muscle weakness & neuropathies showing variable progression to seizures, gait disturbances & paralysis before being non-responsive.
MLD may present at different ages with variable clinical features as described below:
Late-Infantile form (LIF): represent more than half the cases with loss of speech that has already developed, loss of muscle tone, development of rigidity, gait disturbance and rapid-progression to non-responsiveness or death during childhood mainly due to aspiration or bronchopneumonia and respiratory failure[1].
Juvenile-form (JF):represents nearly one-third cases presenting in form of behavioral disturbances or poor school performance during childhood to adolescence with slower progression relative to LIF and survival up to third decade of life.
Adult form (AF): represents less than one-third cases and is characterised by psychiatric disturbances like delusions / hallucinations in addition to features seen in JF with periods of waxing & waning. Survival may extend up to 2 - 3 decades after the diagnosis.
Besides involving nervous system, other organs most commonly gall bladder may be involved with MLD. Few previous case reports have described enlargement of gall bladder with thickened walls, pericholecystic fluid and peripheral calcifications in a case of MLD[6]. Brain MRI in classical case reveals bilaterally, symmetrical, homogeneous & confluent hyperintensity on both T2 weighted & T2FLAIR images involving primarily periventricular white matter involving all cerebral lobes in posteroanterior pattern with extension in to corpus callosum & relative sparing of subcortical white matter in early stages[7]. The dysmyelination occurs typically in tigroid or leopard-skin pattern in the centrum semiovale due to relative sparing of myelin along transmedullary vessels as seen in our index case[8]. Diffusion- weighted images may also show hyperintensities in the corresponding region in the active stage without significant abnormality on ADC map[9]. Proton MR-Spectroscopy in the areas of hyperintensity shows increased Cho/Cr & Cho/NAA ratios and decreased NAA/Cr ratio along with myoinositol peak[10]. Besides, the central nervous system, peripheral nervous system may be involved in form of peripheral neuropathy with reduced motor & sensory nerve conduction velocities leading to gait disturbances[11]. Secondary to suggestion of MRI diagnosis of MLD, low ARSA enzyme activity in leukocytes or chromosomal analysis for ARSA gene may be used for final confirmation[12,13].
Till date, there is no definitive treatment for MLD and management is limited to palliative & supportive measures including gastric tube feeding, antibiotic therapy and bone marrow replacement. Hematopoietic stem cell transplant has shown to delay the progress of disease especially in juvenile form[14,15].
Conclusion
To summarize, though MLD is an uncommon neurological entity yet its characteristic features on MRI brain should prompt us to make an early suggestion of diagnosis so that ARSA activity can be advised in blood leukocytes for final confirmation.
Acknowledge:
I am extremely thankful to MRI technologists viz. Mr. Francis Randhawa & Mr. Hanook Lawrence who took tiring efforts in acquiring and achiving of MR images.
References
- 1. Patil, S.A., Maegawa, G.H. Developing therapeutic approaches for metachromatic leukodystrophy. (2013) Drug Des Devel Ther 7: 729–745.
- 2. Aqarwal, A., Shipman, P.J. Gallbladder polyposis in metachromatic leukodystrophy. (2013) Pediatr Radiol 43(5): 631–633.
- 3. Liaw, H.R., Lee, H.F., Chi, C.S., et al. Late infantile metachromatic leukodystrophy: Clinical manifestations of five Taiwanese patients and Genetic features in Asia. (2015) Orphanet J Rare Dis 10: 144.
- 4. Luzi, P., Rafi, M.A., Rao, H.Z., et al. Sixteen novel mutations in the arylsulfatase A gene causing metachromatic leukodystrophy. (2013) Gene 530(2): 323–328.
- 5. Alessandri, M.G., De Vito, G., Fornai, F. Increased prevalence of pervasive developmental disorders in children with slight arylsulfatase A deficiency. (2002) Brain Dev 24(7): 688–692.
- 6. Rodriguez-Waitkus, P.M., Byrd, R., Hicks, J. Metachromatic leukodystrophy and its effects on the gallbladder: a case report. (2011) Ultrastruct Pathol 35(6): 271-276.
- 7. Martin, A., Sevin, C., Lazarus, C., et al. Toward a better understanding of brain lesions during metachromatic leukodystrophy evolution. (2012) AJNR Am J Neuroradiol 33(9): 1731–1739.
- 8. Nandhagopal, R., Krishnamoorthy, S.G. Neurological picture. Tigroid and leopard skin pattern of dysmyelination in metachromatic leukodystrophy. (2006) J Neurol Neurosurg Psychiatry 77(3): 344.
- 9. Patay, Z. Diffusion-weighted MR imaging in leukodystrophies. (2005) Eur Radiol 15(11): 2284-2303.
- 10. Abdelsalam, E.M., Ashamallah, G.A., Lateef, M.A., et al. Proton MR Spectroscopy in leukodystrophies. (2015) The Egyptian Journal of Radiology and Nuclear Medicine 46 (4): 1091-1097.
- 11. Bindu, P.S., Mahadevan, A., Taly, A.B., et al. Peripheral neuropathy in metachromatic leukodystrophy. A study of 40 cases from south India. (2005) J Neurol Neurosurg Psychiatry 76(12): 1698–1701.
- 12. Kohlschütter, A., Eichler, F. Childhood leukodystrophies: a clinical perspective. (2011) Expert Rev Neurother 11(10): 1485–1196.
- 13. Wang, R.Y., Bodamer, O.A., Watson, M.S., et al. Lysosomal storage diseases: diagnostic confirmation and management of presymptomatic individuals. (2011) Genet Med 13(5): 457–484.
- 14. Krageloh-Mann, I., Groeschel, S., Kehrer, C., et al. Juvenile metachromatic leukodystrophy 10 years post transplant compared with a non-transplanted cohort. (2013) Bone Marrow Transplantation 48(3): 369–375.
- 15. Boucher, A.A., Miller, W., Shanley, R., et al. Long-term outcomes after allogeneic hematopoietic stem cell transplantation for metachromatic leukodystrophy: the largest single-institution cohort report. (2015) Orphanet J Rare Dis 10: 94.










