
Novel Mechanisms of Action for Immunomodulatory Drugs (IMiDs) Against Multiple Myeloma: From a Tragedy to a Therapy
Fengyan Jin1*, Wei Li1, Joel G. Turner2, Yun Dai1,3*
Affiliation
- 1Cancer Center, The First Hospital of Jilin University, Changchun, Jilin, China
- 2Chemical Biology and Molecular Medicine Program, H. Lee Moffitt Cancer Center and Research Institute, Tampa, FL, USA
- 3Division of Hematology and Oncology, Department of Medicine, Virginia Commonwealth University and the Massey Cancer Center, Richmond, VA 23298, USA
Corresponding Author
Fengyan Jin and, Cancer Center, the First Hospital of Jilin University, 71 Xinmin Street, Changchun, Jilin 130021, China, Tel: +8643188782054; Fax: +8643188786102; E-mail: Fengyanjin@jlu.edu.cn; Yun Dai, Virginia Commonwealth University and the Massey Cancer Center, Room 234 Goodwin Research Building, 401 College Street, Richmond VA 23298, Tel: +804-828-5168; Fax: +804-827-3781; E-mail: ydai@vcu.edu
Citation
Jin, F., et al. Novel Mechanisms of Action for Immunomodulatory Drugs Against Multiple Myeloma: from a Tragedy to a Therapy (2016) Int J Hematol Therap 2(1): 1-6.
Copy rights
© 2015 Dai, Y. This is an Open access article distributed under the terms of Creative Commons Attribution 4.0 International License.
Keywords
IMiD; Mechanism of action; Cereblon; IKZF; Multiple myeloma
Abstract
Thalidomide, a sedative to ease morning sickness in pregnant women, was banned more than a half century ago due to causing a severe birth defect known as phocomelia. However, the late discovery of its immunomodulatory properties have led to a new era in the treatment of Multiple Myeloma (MM), a fatal disease that back then had no effective therapy available. The recent development and FDA-accelerated approval of the second- and third-generation thalidomide analogs lenalidomide and pomalidomide to treat MM have led to a paradigm shift in standard of care of MM patients by introducing this novel class of anti-MM therapeutics, termed Immunomodulatory Drugs (IMiDs). Although IMiDs have tremendously improved survival of MM patients, their Mechanism Of Action (MOA) remains largely unknown until cereblon, a component of the E3 ubiquitin ligase complex CRL4CRBN, was discovered as a primary target of IMiDs for both thalidomide teratogenicity and IMiD anti-MM activity. Furthermore, studies have also identified IKZF1 and IKZF3 as the downstream substrates and effectors of cereblon to mediate dual actions of IMiDs, targeting both the MM bone marrow microenvironment and MM cells themselves. These cutting-edge findings have not only tackled the long-lasting conundrum for MOAs of IMiDs but more importantly could lead to more precise use of these agents in the treatment of MM. This article summarizes the new insights into the MOAs of IMiDs as an anti-MM therapy, with a perspective on the existing issues and future directions in this field.
Introduction
Multiple Myeloma (MM) is a malignant disease of mature plasma cells, most frequently diagnosed among elderly people aged 65 to 74 years. In the United States, it is estimated that, in 2015, MM afflicts approximately 26,850 new patients per year[1], accounting for 1.6% of all new cancer cases (ranked at the 14th among common types of cancer), as well as causes about 11,240 deaths per year, comprising 1.9% of all cancer deaths[2]. MM also contributes 15% of the total number of hematologic malignancies[3]. Approximately 86,000 cases per year are newly diagnosed as MM worldwide[4]. MM is characterized by accumulation of malignant plasma cells in bone marrow, accompanied by high levels of kappa or lambda light chain protein in peripheral blood. Over the past decade, the discovery, development, and approval of immunomodulatory agents (IMiDs, e.g., thalidomide [Thalomid®], lenalidomide [Revlimid®], and pomalidomide [Pomalyst®][5] and proteasome inhibitors (e.g., bortezomib [Velcade®] and carfilzomib [Kyprolis®][6] have resulted in a revolution in the treatment of MM, which have significantly increased both remission rate and depth in MM patients. Together with transplants[7], the introduction of these novel agents into the treatment armamentarium of MM has prolonged the median survival of patients from 2 to 3 years to at least 7 years[8]. Therefore, IMiDs and proteasome inhibitors represent two milestones in the field of MM treatment.
IMiDs act both to modulate immune responses and to directly kill MM cells by targeting interactions between MM cells and their bone marrow microenvironment[9]. Whereas the development of IMiDs represents a remarkable example of rapid bench-to-bedside translation of targeted therapies in MM[10], the actual target and mechanism of action for these agents remain unclear until the recent discovery of cereblon, a subunit of an E3 ubiquitin ligase complex, as a primary target of IMiDs, including thalidomide, lenalidomide, and pomalidomide. In this article, we review novel state-of-the-art insights into the Mechanisms Of Action (MOAs) for IMiDs to treat MM.
MM and the Bone Marrow Microenvironment
A distinct feature of MM cells is the requirement for an intimate relation with the bone marrow microenvironment, where malignant plasma cells are nurtured in specialized niches that maintain their long-term survival and protect them from drug-induced apoptosis[11]. During myeloma development and disease progression, the bone marrow microenvironment plays a critical role in virtually all pathogenic aspects of this disease, such as survival, growth, differentiation, invasion/metastasis, and drug resistance of MM cells. The bone marrow microenvironment is composed of various noncellular extracellular matrix components and five major types of stromal cells, including fibroblast-like stromal cells, osteoblasts, osteoclasts, vascular endothelial cells, and lymphocytes[12]. In the former, MM cells directly interact with extracellular matrix components (e.g., fibronectin, collagens, laminin, vitronectin) via integrin- or heparin sulfate proteoglycans (e.g., CD138)-mediated adhesion, as well as with hyaluronan via CD44 isoforms (e.g., CD44v9, CD44v6), RHAMM (CD168), and possibly CD38[13]. Although such interactions with the extracellular matrix promotes survival, proliferation, and migration of MM cells through activation of the signaling pathways (e.g., Src/FAK) in MM cells, one of the most important consequences is cell adhesion-mediated drug resistance[14], a major mechanism underlying drug resistance in MM[15]. In the latter, although MM cells can physically contact with certain types of stromal cells (e.g., via the SDF-1/CXCR4 axis or binding of vascular cell adhesion molecule-1 with VLA4), bone marrow stromal cells act primarily to secret, via paracrine, cytokine, and growth factors. These include interleukin-6 (IL-6), insulin-like growth factor 1, basic fibroblast growth factor, hepatocyte growth factor, vascular endothelial growth factor, tumor necrosis factor α (TNFα), B-cell activating factor, a proliferation-inducing ligand (APRIL), and stromal cell-derived factor-1α (SDF-1α, also known as C-X-C motif chemokine 12/CXCL12)[16]. These factors bind to their corresponding receptors on MM cells and thus activate multiple signaling pathways (e.g., MEK/ERK, PI3K/Akt or /PKC, JAK/Stat3, NF-αB) essential for survival, proliferation, invasion/metastasis, and drug resistance of MM cells and also critical for MM-related angiogenesis and bone destruction[17]. In return, MM cells produce a large amount of immunosuppressive factors (e.g., TGFα, IL-10, IL-6), resulting in immune tolerance, a mechanism by which MM cells evade immune surveillance through inhibiting the function of immune cells, such as dendritic cells, natural killer cells, T cells, and B cells, while activating regulatory T cells[17,18]. Meanwhile, MM cells also secret macrophage inflammatory protein-1α that stimulates osteoclastogenesis, as well as IL-3 and Dickkopf 1 (DKK1) that inhibit osteoblastogenesis, together promoting osteolysis in MM-related bone disease[16]. Therefore, approaches targeting the interactions between MM cells and the bone marrow microenvironment have attracted tremendous attention in the treatment of MM, among which IMiDs represent a paradigm.
Development of IMiDs with Dual Actions via Targeting the MM Microenvironment
Thalidomide, a first-generation IMiD, was first marketed in the late 1950s by the West Germany pharmaceutical company Chemie Grunenthal GmbH. It was prescribed as a sleeping pill (or tranquilizer) and particularly used as an antiemetic and sedative to ease morning sickness in pregnant women. In 1960, thalidomide was found to cause a severe birth defect known as phocomelia, a name for the flipper-like limbs, and almost immediately withdrawn from the market due to this teratogenic adverse effect in 1961. However, anti-inflammatory properties of thalidomide were later discovered that improved the clinical signs and symptoms in patients with erythema nodosum leprosum. The mechanism for this effect remained unknown until the early 1990s when this agent was found to inhibit production of tumor necrosis factor-(TNFα), a major cytokine that mediates inflammation. Based on these findings, thalidomide has thus been used to treat certain inflammatory diseases such as erythema nodosum leprosum, AIDS-related cachexia, and aphthous ulcers, as well as autoimmune diseases (e.g., chronic graft versus host disease and Crohn disease), thereby named as an immunomodulatory drug (IMiD).
In 1994, Folkman, an American scientist who first reported anti-angiogenesis as a potential anti-cancer therapy due to dependency of solid tumors on angiogenesis induced by a factor secreted from these tumors, and his colleagues discovered that thalidomide functions as an inhibitor of angiogenesis triggered by basic fibroblast growth factor in a rabbit cornea micropocket assay[19]. These findings on the anti-angiogenic property of thalidomide have provided a new insight into the mechanism for thalidomide teratogenicity and also raise the possibility of using this agent to treat many angiogenesis-dependent diseases including cancer. Among several malignancies tested for the anti-angiogenic activity of thalidomide, a research group at the University of Arkansas first reported the activity of thalidomide as a single agent in MM patients who had been previously heavily treated[20]. It was then further found that thalidomide synergistically interacts with dexamethasone in MM. In 1998, the treatment of MM entered an entirely new era, landmarked with the approval of thalidomide (in combination with dexamethasone) by the US Food and Drug Administration (FDA) in the treatment of patients with newly diagnosed MM. Among numerous clinical studies of thalidomide in MM, the most important one was conducted by Singhal et al.[21], in which 84 patients with relapsed and refractory MM orally received an initial dose of 200 mg daily, followed by a step-wise increase of 200 mg per day every 2 weeks until patients reached the maximum daily dose of 800 mg. Using serum and urine light chain protein as therapeutic markers for an objective response, the group found that the overall response rate was 32%, with 1-year overall survival rate of 58% and event-free survival rate of 22%, respectively. Of note, 10% of patients achieved a complete or near-complete response or remission. To some degree, the patients who had been heavily treated with high-dose chemotherapy also responded. Although this study consolidates the position of thalidomide in the treatment of MM, such high doses of thalidomide were not well tolerated. Severe adverse events, together with other complications (e.g., poor water solubility, poor absorption in the intestines), have promoted the development of thalidomide analogs with fewer side effects and increased potency.
With modification of the chemical structure of thalidomide (e.g., adding an amino group at position 4 and removing a carbonyl group in the phthaloyl ring), two of the new analogs, amino-EM-12 and amino-thalidomide, have been found to be more bioavailable and more potent in inhibition of TNF α production. These two agents were later approved by the FDA as lenalidomide (along with dexamethasone) to treat patients with MM who have received one prior therapy and pomalidomide to treat patients who have received at least two prior therapies in 2006[22] and 2013[5], respectively. Thus, these represent the second- and third-generation IMiDs in the treatment of MM.
Whereas IMiDs have displayed remarkable effectiveness against MM, numerous studies have been carried out in order to understand their MOAs. In addition to the original functions that target MM bone marrow microenvironment (e.g., inhibition of TNF α production, through PDE4-dependent or independent process, and angiogenesis)[9], it has been found that IMiDs also act directly against MM cells. For example, exposure to thalidomide arrests MM cells at G1 phase by up-regulation of p21Cip1, an endogenous Cdk inhibitor, as well as induces cell death in both MM cell lines and primary CD138+ MM cells[23]. IMiDs, with their immunomodulatory properties, can also kill MM cells indirectly by stimulating T cells to secret IL-2 and increasing cytotoxic activity of natural killer cells[24]. Again, IMiDs synergistically interact with dexamethasone to kill MM cells, which is consistent with numerous clinical findings[25]. More recently, anti-MM activity of IMiDs has also been related to alterations in epigenetic status of MM cells through targeting of the bone marrow microenvironment[11].
Discovery of Cereblon as a Primary Target of IMiDs
While the dual actions of IMiDs against MM have been well documented, the MOAs of thalidomide and its analogs remain not fully understood until their molecular target has been discovered very recently. In 2010, Ito et al identified cereblon as a thalidomide-binding protein[26], which forms an E3 ubiquitin ligase complex (termed CRL4CRBN) with DNA damage-binding protein 1 (DDB1), CUL4A, and ROC1[27]. The crystal structure of human cereblon bound to DDB1 and lenalidomide later was revealed as the basis for direct binding between IMiDs and cereblon-DDB1[28] and also characterizes the key drug-binding residues responsible for anti-MM effects of IMiDs[29]. Binding of thalidomide inhibits cereblon auto-ubiquitination and E3 ubiquitin ligase activity of the cereblon-containing complex, resulting in severely reduced expression of fibroblast growth factor 8, a secretory factor critical for limb outgrowth during embryo development. As a consequence, these events lead to thalidomide-induced developmental defects in both zebrafish and chicks, which can be reversed by blocking thalidomide-cereblon interaction using point mutations of cereblon (e.g., CRBNY384A/W386A)[26]. Therefore, cereblon has been identified as a primary target for thalidomide to cause teratogenicity, a mystery that has lasted over a half century.
The finding of cereblon as a primary target of thalidomide in teratogenicity has rapidly transformed into an understanding of the MOAs underlying the anti-MM activity of IMiDs. The most important findings include (1) like thalidomide, other IMiDs, such as lenalidomide and pomalidomide, also bind to and inhibit auto-ubiquitination of cereblon within the cereblon-DDB1 complex[30] and (2) siRNA knockdown of cereblon reduces viability of MM cells, while residual living cells with lower levels of cereblon become highly resistant toward lenalidomide and pomalidomide. Moreover, loss of cereblon is associated with acquired lenalidomide or pomalidomide resistance in human MM cells lines[31]. Further findings include (3) whereas overexpression of cereblon sensitizes MM cell to IMiDs[29], siRNA knockdown of the cereblon gene results in lenalidomide resistance[30,31] and (4) consistent with in vitro results described above, patients with MM highly expressing cereblon respond better to IMiDs[32], whereas a lower cereblon level is common in lenalidomide refractory patients[33]. The conclusion is that cereblon is an essential target required for anti-MM activity of IMiDs and may also represent a biomarker to predict response of MM patients to these agents.
Next, interferon regulatory factor 4 (IRF4) has been identified as a downstream target of cereblon involving the MOAs of IMiDs. It has been found that, whereas treatment with IMiDs downregulates expression of IRF4, knockdown of IRF4 promotes apoptosis of tumor cells, indicating its critical function for survival of MM and B-cell-like subtype diffuse large B-cell lymphoma cells[31,34]. Moreover, IRF4 can induce expression of MYC, a well-known oncogene highly expressed in MM cells, rather than normal plasma cells[35]. (Table 1) Because MYC plays important roles in tumor cell survival and disease progression in MM, it has been confirmed that IMiDs targets cereblon to directly kill MM and lymphoma cells through downregulation of MYC due to IRF4 inhibition[36,37]. On the other hand, IMiDs also target cereblon to inhibit production TNFα and IL-2[38], providing a plausible explanation for their immunomodulatory properties. Together, discovery of cereblon as a thalidomide target uncovers the dual actions of IMiDs in the treatment of MM[39].
Table 1: Different Downstream Molecules of Cereblon Responsible for Varied Effects of Imids
| IMiD | Downstream Target | Effect | |
|---|---|---|---|
| Phocomelia | Tha | FGF8 | Limb outgrowth defect |
| MM | Tha/Len/Pom | IKZF1/3→IRF4→MYC | Killing/inhibiting MM cells |
| IKZF1/3→IRF4→IL-2 | Immunomodulatory action | ||
| MDS | Len | CK1α | Killing/inhibiting MDS cells with del(5q) |
Tha, thalidomide; Len, lenalidomide; Pom, pomalidomide
Finally, two DNA-binding transcription factors, Ikaros family zinc finger protein 1 (IKZF1, also known as Ikaros) and 3 (IKZF3, also known as Aiolos), have been discovered as the last missing piece of the puzzle for the cereblon-mediated dual actions of IMiDs against MM. In 2014, two groups have simultaneously identified IKZF1 and IKZF3 as the substrates of cereblon, whereas direct binding of IMiDs to cereblon results in ubiquitination and subsequent proteasomal degradation of these two proteins[36,38]. IKZF1 and IKZF3 are members of the Ikaros family containing zinc finger structure, which play crucial roles in differentiation and development of lymphocytes as well as survival of MM cells. IKZF1 binds to and activates the promoter of IRF4, whereas IRF4 upregulation in turn activates its downstream gene MYC, therefore promoting survival and proliferation of MM cells. Conversely, ubiquitination and degradation of IKZF1 due to binding of IMiDs to cereblon result in downregulation of IRF4 and thusMYC, leading to direct killing and inhibiting growth of MM cells by IMiDs[39]. However, it is noteworthy that lenalidomide also inhibits proliferation of MM cells with high basal levels of IRF4 but does not downregulate IRF4, suggesting other unknown downstream targets of the cereblon/IKZF signal. On the other hand, IKZF3 functions to suppress the promoter of the IL-2 gene. Therefore, degradation of IKZF1/3 by IMiDs (e.g., lenalidomide, pomalidomide) through binding to cereblon releases this transcriptional repression, thus promoting production of IL-2 by T cells[38,40], which in turn mediates the immunomodulatory action of these agents in the treatment of MM. Moreover, it has been demonstrated that MM patients with lower levels of IKZF1 have a worse response and shorter survival after treatment with IMiDs[33]. Together, as summarized in Figure 1, these very recent findings on the role of these cereblon/IKZF-mediated events provide a cutting-edge insight into the MOAs underlying the dual anti-MM actions of IMiDs. They also lay a basis for discovery of new biomarkers for selection of eligible patients and prediction of therapeutic response to IMiDs, as well as for future research and development of new agents and combination strategies targeting these novel pathways to improve efficacy of IMiDs in the treatment of MM.
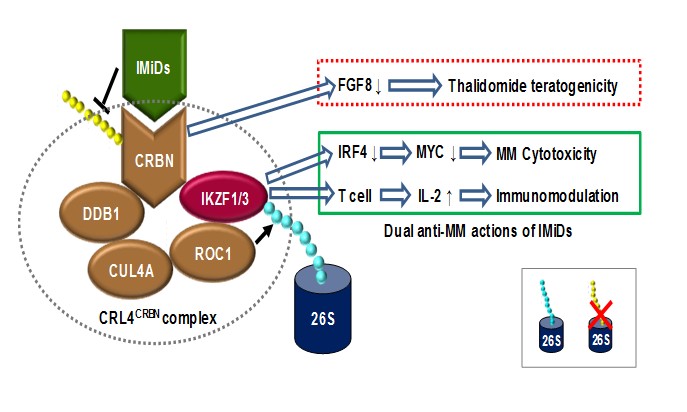
Figure 1: Mechanisms Of Action (MOAs) for the dual actions of IMiDs against MM. Cereblon (CRBN), a subunit of the CRL4CRBN complex consisting of cereblon, DDB1 (DNA damage-binding protein 1), CUL4A (cullin 4A), and ROC1 (regulator of cullins 1), serves as the primary target of immunomodulatory drugs (e.g., thalidomide, lenalidomide, and pomalidomide). Binding of thalidomide to cereblon prevents DDB1- and CUL4A-dependent auto-ubiquitination, inhibiting its associated ubiquitin ligase activity, resulting in downregulation of fibroblast growth factor 8, a critical secretory factor for limb outgrowth during embryo development. As consequence, exposure of pregnant women to thalidomide causes the birth defect known as phocomelia, a name for the flipper-like limbs. However, binding of IMiDs to cereblon specifically recruits 2 members of the Ikaros family zinc finger transcription factors, IKZF1 (Ikaros) and IKZF3 (Aiolos), to the CRL4CRBN complex, resulting in their ubiquitination and subsequent proteasomal degradation. Whereas the Ikaros transcription factors are essential for regulating gene expression during development of lymphocytes, IKZF1 and IKZF3 also play critical roles in tumor cell survival of B-cell malignancies, especially MM, as well as in transcriptional repression of IL-2 expression in T cells. Therefore, downregulation of IKZF1/3 via a CRBN-dependent process provides a mechanism for cytotoxicity of IMiDs directly toward MM cells, via inhibiting expression of IRF4 and thus its target MYC. On the other hand, cereblon-dependent downregulation of IKZF1/3 also releases their repression of IL-2 expression in T cells, resulting IL-2 production that in turn mediates immunomodulatory effects of IMiDs. Together, these cereblon/IKZF-associated events shed light into understanding the MOAs responsible for the dual actions of IMiDs in the treatment of MM.
Perspectives
The current breakthrough discoveries have uncovered the long-lasting truth for MOAs of thalidomide and its analogs in teratogenicity and in treatment of MM. They may also provide a foundation for discovering biomarkers and therapeutic targets involving the cereblon/IKZF-mediated signaling pathways, potentially leading to more targeted medicine using IMiDs to treat patients with MM. However, recent studies also raise a possibility that disruption of the cereblon-IKZF-IRF4 cascade is not the whole story to interpret the dual actions of IMiDs against MM in the clinical settings, particularly regarding why a number of MM patients do not respond or become resistant to IMiDs.
First, unlike the proteasome inhibitors, another landmark in the field of MM treatment, IMiDs by themselves are often not very potent in rapidly killing MM cells, at least in vitro in MM cell lines. Similarly, direct downregulation of MYC, known as a major downstream effect or of the cereblon/IKZF/IRF4 cascade, by other approaches (e.g., JQ1, a potent inhibitor of the BET family of bromodomain proteins) does not effectively induce apoptosis of MM cells in vitro and rather markedly inhibits cell growth due to cell cycle arrest and senescence[41]. This reality highlights the predo; minant role of another cereblon-mediated MOA (i.e., immunomodulatory effects that targets MM bone marrow microenvironment) in the effectiveness of IMiDs toward MM.
Second, inhibition of IKZF1 and IKZF3 proteasomal degradation trigged by cereblon-mediated ubiquitination should theoretically impair or antagonize the anti-MM capacity of IMiDs. However, it is not true as a high-degree synergy between the proteasome inhibitor bortezomib and lenalidomide has been well documented, which in fact provides a foundation for the current first-line regimen combining these two agents in the treatment of high risk MM[42]. To this end, synergistic interactions between IMiDs and proteasome inhibitors seem to occur independently of the cereblon/IKZF-mediated process. Thus, there exist other unknown MOAs of IMiDs for this clinically significant combination regimen.
Third, IKZF1 mutation/deletion is associated to oncogenic tyrosine kinase activation in acute lymphoid leukemia (ALL) and represents an independent factor for poor prognosis of patients with ALL[43]. The opposite functions of IKZF1 in ALL (tumor-suppressor) versus MM (oncoprotein) raises a possibility that there may be different isoforms of IKZF1 with distinct functions in these two kinds of hematological malignancies. They also raise a question regarding whether IMIDs such as lenalidomide would promote growth of pre-B tumor cells in ALL.
Fourth, although IMIDs bind to cereblon as the primary target, the downstream substrates of cereblon may vary between different diseases. In this context, while IMiDs induce ubiquitination and degradation of IKZF1 and IKZF3 in MM cells, lenalidomide acts to induce ubiquitination and degradation of another cereblon substrate, casein kinase 1 (CK1α)[44], via the CUL4-RBX1-DDB1-cereblon complex (CRL4CRBN)[45], in a subtype of myelodysplastic syndrome (MDS) characterized by deletion of chromosome 5q (del(5q))[46]. Of note, lenalidomide is also highly active in this specific del(5q) MDS subtype[47]. Not surprising, CK1α is encoded by a gene, CSNK1A1, that localizes within the common deleted region for del(5q) MDS. In striking contrast, thalidomide and CC-122, a novel CRBN-binding agent, have no effect on CK1α protein levels. Interestingly, lenalidomide is the only one IMiD that is effective against del(5q) MDS in clinic. Consistently, a single deuterated enantiomer related to CC-122 displays a profoundly different potency in responses of human MM cells[48]. Thus, conformational flexibility and structural dynamics of cereblon, as observed via X-ray crystallography[49], might result in significant changes on the surface of cereblon, near the IMID-binding site, which is probably responsible for the effects of IMiDs on different cereblon substrates, such as IKZF1 versus CK1α.
Fifth, whereas approximately 80% of MM patients who respond to lenalidomide display high basal levels of cereblon[50], there is however no difference in expression of IKZF1/3 between responders and nonresponders to IMiDs[50]. There is also no correlation between the level of cereblon, IKZF1, and IKZF3 proteins in primary CD138+ MM cells[50], indicating the complexity of IMiD MOA. While basal levels of cereblon and/or IKZF do not always correlate to better response to IMiDs, it remains to be defined whether either or both of them can serve as an independent biomarker to predict who would most likely benefits from IMiD treatment.
Last, although lower levels of cereblon or IKZF is, at least in part, associated with a worse response to IMiDs, there is no report demonstrating whether any aberration (e.g., mutation, amplification, deletion, rearrangement, etc.) of these genes is related to intrinsic or acquired resistance to IMiDs. For example, whereas manipulation of the cereblon gene at thalidomide-binding sites (e.g., mutations of Y384, W386, or both sites) significantly impairs biological effects of thalidomide[30], these mutations are so far not found in MM patients who fail to respond to IMiDs. Therefore, it remains virtually unknown whether any genetic abnormalities of cereblon, IKZFs, or other related genes contribute to IMiD resistance in MM.
In conclusion, whereas the recent advances in understanding MOAs of IMiDs are revolutionary, particularly in the field of MM treatment, the issues stated above as well as some others warrant considerations in translation of these cutting-edge findings into clinical practice.
Acknowledgment
This work was supported by National Natural Science Foundation of China (Grant No. 81471165), Jilin Province Health Family Planning Research Plan (Grant No. 20142041), the Program of Jilin Provincial Science & Technology Department (Grant No. 20150520143JH), and the Young Scholar Program of Norman Bethune Health Science Center of Jilin University (Grant No. 2013206044).
Conflict of interest
No potential conflicts of interest were disclosed.
References
- 1. Siegel, R., Naishadham, D., Jemal, A. Cancer statistics. (2013) CA Cancer J Clin 63: 11-30.
- 2. SEER Stat Fact Sheets: Myeloma.
- 3. Jagannath, S., Richardson, P.G., Munshi, N.C. Multiple myeloma and other plasma cell dyscrasias. (2014) Cancer Management 1-22.
- 4. Moreau, P., Attal, M., Facon, T. Frontline therapy of multiple myeloma. (2015) Blood 125: 3076-3084.
- 5. Lacy, M.Q., McCurdy, A.R. Pomalidomide. (2013) Blood 122(14): 2305-2309.
- 6. Herndon, T., Deisseroth, A.B., Kaminskas,E., et al. U.S. Food and Drug Administration Approval: Carfilzomib for the treatment of multiple myeloma. (2013) Clin Cancer Res 19: 4559-4563.
- 7. Bekadja, M.A., Bouhass, R. Non-cryopreserved periperal stem cell (pscs) Autograft for multiple myeloma and lymphoma in developing countries. (2015) Int J Hematol Therap 1: 1-6.
- 8. Ocio, E.M., Mitsiades, C.S., Orlowski, R., et al. Future agents and treatment directions in multiple myeloma. (2014) Expert Rev Hematol 7: 127-141.
- 9. Anderson, K.C. Lenalidomide and thalidomide: mechanisms of action - similarities and differences. (2005) Semin Hematol 42: S3-8.
- 10. Anderson, K.C. The 39th David A. Karnofsky Lecture: bench-to-bedside translation of targeted therapies in multiple myeloma. (2012) J Clin Oncol 30: 445-452.
- 11. Boyle, E.M., Davies, F.E., Leleu, X., et al. Understanding the multiple biological aspects leading to myeloma. (2014) Haematologica 99(4): 605-612.
- 12. Mimura, N., Hideshima, T., Anderson, K.C. Novel therapeutic strategies for multiple myeloma. (2015) Exp Hematol 43(8): 732-741.
- 13. Katz, B.Z. Adhesion molecules – The lifelines of multiple myeloma cells. (2010) Semin Cancer Biol 20(3): 186-195.
- 14. Hazlehurst, L.A., Landowski, T.H., Dalton, W.S. Role of the tumor microenvironment in mediating de novo resistance to drugs and physiological mediators of cell death. (2003) Oncogene 22(47): 7396-7402.
- 15. Neri, P., Bahlis, N.J. Targeting of adhesion molecules as a therapeutic strategy in multiple myeloma. (2012) Curr Cancer Drug Targets 12(7): 776-796.
- 16. Hideshima, T., Mitsiades, C., Tonon, G., et al. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. (2007) Nat Rev Cancer 7(8): 585-598.
- 17. Pagnucco, G., Cardinale, G., Gervasi, F. Targeting multiple myeloma cells and their bone marrow microenvironment. (2004) Ann N Y Acad Sci 1028: 390-399.
- 18. Quach, H., Ritchie, D., Stewart, A.K., et al. Mechanism of action of immunomodulatory drugs (IMiDs) in multiple myeloma. (2010) Leukemia 24(1): 22-32.
- 19. D’Amato, R.J., Loughnan, M.S., Flynn, E., et al. Thalidomide is an inhibitor of angiogenesis. (1994) Proc Natl Acad Sci U.S.A. 91(9): 4082-4085.
- 20. Rhee, F., Dhodapkar, M., Shaughnessy, Jr. J.D., et al. First thalidomide clinical trial in multiple myeloma: a decade. (2008) Blood 112(4): 1035-1038.
- 21. Singhal, S., Mehta, J., Desikan, R., et al. Antitumor activity of thalidomide in refractory multiple myeloma. (1999) N Engl J Med 341: 1565-1571.
- 22. Scott, L.J., Lyseng-Williamson, K.A. Lenalidomide: A review of its use in the treatment of relapsed or refractory multiple myeloma. (2011) Drugs 71(5): 625-649.
- 23. Hideshima, T., Chauhan, D., Shima, Y., et al. Thalidomide and its analogues overcome drug resistance of human multiple myeloma cell lines to conventional therapy. (2000) Blood 96(9): 2943-2950.
- 24. Davies, F.E., Raje, N., Hideshima, T., et al. Thalidomide and immunomodulatory derivatives augment natural killer cell cytotoxicity in multiple myeloma. (2001) Blood 98(1): 210-216.
- 25. Benboubker, L., Dimopoulos, M.A., Dispenzieri, A., et al. Lenalidomide and dexamethasone in transplant-ineligible patients with myeloma. (2014) N Engl J Med 371(10): 906-917.
- 26. Ito, T., Ando, H., Suzuki, T., et al. Identification of a primary target of thalidomide teratogenicity. (2010) Science 327(5971): 1345-1350.
- 27. Lupas, A.N., Zhu, H., Korycinski, M. The thalidomide-binding domain of cereblon defines the CULT domain family and is a new member of the β-tent fold. (2015) PLoS Comp Biol 11(1): e1004023.
- 28. Fischer, E.S., Bohm, K., Lydeard, J.R., et al. Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide. (2014) Nature 512(7512): 49-53.
- 29. Chamberlain, P.P., Lopez-Girona, A., Miller, K., et al. Structure of the human Cereblon–DDB1–lenalidomide complex reveals basis for responsiveness to thalidomide analogs. (2014) Nat Struct Mol Biol 21(9): 803-809.
- 30. Lopez-Girona, A., Mendy, D., Ito, T., et al. Cereblon is a direct protein target for immunomodulatory and antiproliferative activities of lenalidomide and pomalidomide. (2012) Leukemia 26(11): 2326-2635.
- 31. Zhu, Y.X., Braggio, E., Shi, C.X., et al. Cereblon expression is required for the anti-myeloma activity of lenalidomide and pomalidomide. (2011) Blood 118(18): 4771-4779.
- 32. Gandhi, A.K., Waldman, M., Chen, G., et al. Measuring cereblon as a biomarker of response or resistance to lenalidomide and pomalidomide requires use of standardized reagents and understanding of gene complexity. (2014) Br J Haematol 164(2): 233-244.
- 33. Zhu, Y.X., Braggio, E., Shi, C-X., et al. Identification of cereblon-binding proteins and relationship with response and survival after IMiDs in multiple myeloma. (2014) Blood 124(4): 536-545.
- 34. Zhang, L.H., Kosek, J., Wang, M., et al. Lenalidomide efficacy in activated B-cell-like subtype diffuse large B-cell lymphoma is dependent upon IRF4 and cereblon expression. (2013) Br J Haematol 160: 487-502.
- 35. Shaffer, A.L., Emre, N.C., Lamy, L., et al. IRF4 addiction in multiple myeloma. (2008) Nature 454(7201): 226-231.
- 36. Lu, G., Middleton, R.E., Sun, H., et al. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. (2014) Science 343(6168): 305-309.
- 37. Gopalakrishnan, R., Matta, H., Tolani, B., et al. Immunomodulatory drugs target IKZF1-IRF4-MYC axis in primary effusion lymphoma in a cereblon-dependent manner and display synergistic cytotoxicity with BRD4 inhibitors. (2015) Oncogene 245.
- 38. Krönke, J., Narla, A., Udeshi, N.D., et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. (2014) Science 343(6168): 301-305.
- 39. Krönke, J., Hurst, S.N., Ebert, B.L. Lenalidomide induces degradation of IKZF1 and IKZF3. (2014) OncoImmunology 3(7): e941742.
- 40. Gandhi, A.K., Kang, J., Havens, C.G., et al. Immunomodulatory agents lenalidomide and pomalidomide co-stimulate T cells by inducing degradation of T cell repressors Ikaros and Aiolos via modulation of the E3 ubiquitin ligase complex CRL4(CRBN). (2014) Br J Haematol 164(6): 811-821.
- 41. Delmore, J.E, Issa, G.C., Lemieux, M.E., et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. (2011) Cell 146(6): 904-917.
- 42. Weber, D.M., Chen, C., Niesvizky, R., et al. Lenalidomide plus dexamethasone for relapsed multiple myeloma in North America. (2007) N Engl J Med 357(21): 2133-2142.
- 43. Mullighan, C.G., Su, X., Zhang, J., et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. (2009) N Engl J Med 360: 470-480.
- 44. Schneider, R.K., Adema, V., Heckl, D., et al. Role of casein kinase 1A1 in the biology and targeted therapy of del(5q) MDS. (2014) Cancer Cell 26(4): 509-520.
- 45. Hartmann, M.D., Boichenko, I., Coles, M., et al. Thalidomide mimics uridine binding to an aromatic cage in cereblon. (2014) J Struct Biol 188(3): 225-232.
- 46. Kronke, J., Fink, E.C., Hollenbach, P.W., et al. Lenalidomide induces ubiquitination and degradation of CK1a in del(5q) MDS. (2015) Nature 523(7559): 183-188.
- 47. Wei, S., Chen, X., Rocha, K., et al. A critical role for phosphatase haplodeficiency in the selective suppression of deletion 5q MDS by lenalidomide. (2009) Proc Natl Acad Sci U.S.A. 106(31): 12974-12979.
- 48. Jacques, V., Czarnik, A.W., Judgeb, T.M. Differentiation of antiinflammatory and antitumorigenic properties of stabilized enantiomers of thalidomide analogs. (2015) Proc Natl Acad Sci U.S.A 112(12): e1471-e1479.
- 49. Hartmann, M.D., Boichenko, I., Coles, M., et al. Structural dynamics of the cereblon ligand binding domain. (2015) PLoS ONE 10(5): e0128342.
- 50. Huang, S.Y., Lin, C.W., Lin, H.H., et al. Expression of cereblon protein assessed by immunohistochemical staining in myeloma cells is associated with superior response of thalidomide and lenalidomide-based treatment, but not bortezomib-based treatment, in patients with multiple myeloma. (2014) Ann Hematol 93(8): 1371-1380.
- 51. Sehgal, K., Das, R., Zhang, L., et al. Clinical and pharmacodynamic analysis of pomalidomide dosing strategies in myeloma: impact of immune activation and cereblon targets. (2015) Blood 125(26): 4042-4051.










