
Oro-Dental Manifestations and Implications of the Osteolyses
Chetty, M1,3*, Roberts, T1,3, Stephen, L.X.G1,3, Bertie, J.D4, Beighton, P2,3
Affiliation
- 1Faculty of Dentistry, University of the Western Cape, Bellville, Cape Town, South Africa
- 2Division of Human Genetics, Faculty of Health Sciences, University of Cape Town, South Africa
- 3University of the Western Cape/ University of Cape Town Combined Dental Genetics Clinic, Red Cross Childrens Hospital, Cape Town, South Africa
- 4Department of Orthopaedics, Greys Hospital, Pietermaritzburg, South Africa
Corresponding Author
Professor Chetty, M., Faculty of Dentistry, University of the Western Cape/ University of Cape Town Combined Dental Genetics Clinic, Red Cross Childrens Hospital, Cape Town, South Africa, Tel: 021 9373112; E-mail: drmchetty@mweb.co.za
Citation
Chetty, M., et al. Oro-Dental Manifestations and Implications of the Osteolyses. (2017) J Dent Oral Care 3(2): 1- 5.
Copy rights
© 2017 Chetty, M. This is an Open access article distributed under the terms of Creative Commons Attribution 4.0 International License.
Keywords
Dental; Genetic; Osteolysis
Abstract
The Osteolyses are a group of uncommon genetic disorders which are characterized by progressive resorption of bone. This process ultimately results in significant physical handicap with pathological fractures representing an additional complication. The skeleton in the Osteolyses is radiolucent and the manifestations resemble those of Osteogenesis Imperfecta.
A boy with a confirmed diagnosis of Torg-Winchester syndrome attended the metabolic bone clinic at Grey’s Hospital and his clinical and radiological features have recently been published[1]. The dental and craniofacial manifestations of this boy form the subject of this article. These features tend to be overshadowed by the more dramatic skeletal changes in the limbs. Nevertheless, appropriate dental care is an important component of the overall management of this severe condition.
Introduction
The Osteolyses are a group of rare genetic disorders which are characterized by progressive resorption of bone. This process, which is maximal in the articular regions of the limbs, leads to significant physical handicap. Pathological fractures represent an additional complication. The skeleton in the Osteolyses is radiolucent and the manifestations resemble those of Osteogenesis Imperfecta.
The nomenclature and classification of the Osteolyses was initially based upon the anatomical distribution of the affected regions and terms such as ‘carpo-tarsal osteolysis’ and ‘multicentric osteolysis’ came into use. With the delineation of specific osteolysis syndromes, eponyms were also employed. These included ‘Winchester’[2] ,‘Torg’[3] and the conjoined eponym is in current use.
There is a paucity of published information pertaining to the orofacial manifestations of the osteolyses. Course facial features and gum hypertrophy have been mentioned[2,4]. The only article found in the oral and maxillofacial literature detailed the clinical features of the Winchester syndrome in a 40 year old woman with course facial features, underdeveloped maxillary sinuses, a prognathic mandible, several impacted teeth, mild gingivitis and a submucous cleft palate[5]. In the dental context[6], described the clinical features of an Italian child whose ‘teeth were discoloured and said to chip easily’.
In South Africa, over the last 40 years, only 4 persons with Osteolysis have been reported in the literature, three in the first article[7] and one in the second[1]. The oro-dental manifestations of the latter person, a boy of Zulu stock with the Torg-Winchester Osteolysis Syndrome have been investigated in detail. In order to draw the attention of dental colleagues to the implications of this rare disorder, the oro-dental manifestations are described, depicted and described in this article.
Nosology
Winchester et al., (1969)[2] reported two siblings from Puerto Rico who presented with a connective tissue disorder characterized by a short stature, joint contractures, corneal opacities, osteoporosis, carpal-tarsal osteolysis, stiff joints, skin lesions and coarse facies. In the same year, Torg et al., (1969)[3] described 3 siblings with mild to moderate osteoporosis, osteolytic changes of the bones of the hands and feet. Their skin manifestations that resembled Winchester syndrome but with subcutaneous nodules and less severe skeletal involvement.
Three decades later, an entity which resembled the Torg syndrome, but comprised severe osteolysis and osteopenia, painful subcutaneous nodules was documented in the endogenous Arabian population[8-10]. This condition was termed ‘Nodulosis-Arthropathy-Osteolysis’ (NAO).
Zankl et al., (2005)[6] suggested that Winchester syndrome was caused by a homozygous mutation in the active site of Matrix Metalloproteinase2 Gene (MMP2) and Rouzier et al., (2006)[11] recorded a homozygous MMP2 mutation in a patient diagnosed with Winchester syndrome. Thereafter, Zankl et al., (2007)[4] identified two separate mutations in the MMP2 gene which resulted in complete loss of matrix metalloproteinase activity. These mutations were in the MMP2 gene at chromosome 16q13 in persons with Torg syndrome, Winchester syndrome and NAO syndrome. On this basis, they suggested that these disparate disorders represented a continuous clinical spectrum which resulted from intragenic heterogeneity in the MMP2 gene.
As cases accumulated, it became evident that there was considerable clinical overlap between the putative entities and the combined eponym ‘Torg-Winchester syndrome’ [OMIM 259600] came into use with NAO syndrome regarded as an allelic variant. In the Torg-Winchester syndrome the wrists and ankles are often affected and there is eventual dissolution of the carpals and tarsals[12].
Other Osteolyses which were listed in the 2010 revision of the International Nosology of Genetic Skeletal Disorders (Warman et al., 2011), included Familial expansile osteolysis, Progeria (Hutchinson-Gilford type), Hadju-Cheney syndrome and Multicentric carpal-tarsal osteolysis with and without nephropathy. The current nosology revision[13] places the Osteolyses in group 28 and confirms that mutations in MMP2 primarily contribute to the phenotypic presentation of the Torg-Winchester Osteolyses.
Case Report
A boy with a confirmed diagnosis of Torg-Winchester syndrome attends the metabolic bone clinic at Grey’s Hospital[1] The dental and craniofacial manifestations of a boy form the subject of this article.
The boy, aged 11 years, belonging to the Zulu community of KwaZulu Natal presented to the orthopaedic clinic with pain and deformity of both hands (Figure 1) and both legs (Figure 2) at the age of 6 years. His intellect and general development were normal. Deformities of both his hands and knees progressed and he became chair bound. His parents and siblings were unaffected and no relative had a similar disorder. The results of routine and specific biochemical and haematological investigations were normal.
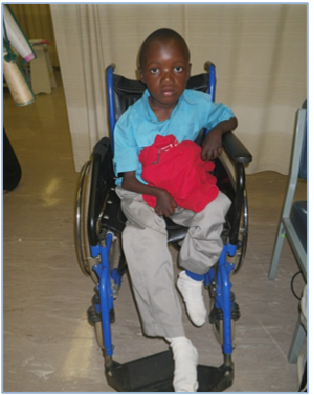
Figure 1: Deformities of both wrists and hands.
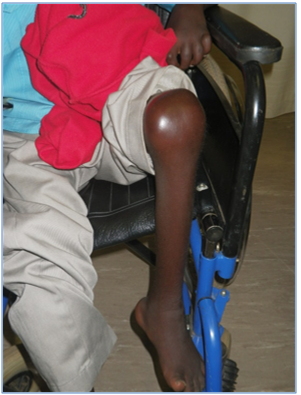
Figure 2: The left knee is swollen and tender.
Radiographs of his hands disclosed an absence of carpal bones of the right wrist joint and osteolysis of the distal region right ulna (Figure 3). Images of his feet revealed deformities of the tarsus and widened lucent metatarsals (Figure 4).
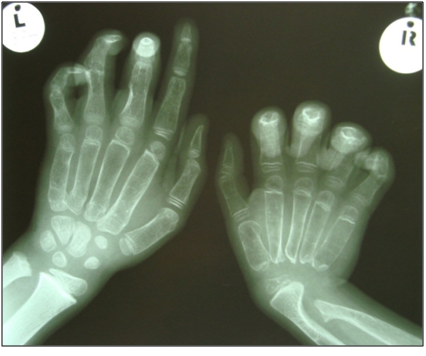
Figure 3: Absence of the carpal bones in the right wrist with widened lucent metacarpals and osteolysis of distal ulna.
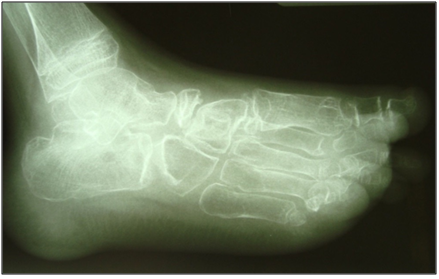
Figure 4: Diffuse osteopaenia of the foot with deformity of the tarsus and widened metatarsals.
Craniofacial findings
Clinical assessment of his craniofacial features revealed frontal bossing, a flattened nasal bridge, ocular hypertelorism and normal sclerae. An intraoral examination was completed with difficulty due to the tense musculature and pain in the region of the Temporomandibular Joint (TMJ) during opening of his mouth. His maximum oral opening was 20 mm. Moderate gingivitis was evident but there were no obvious carious or periodontal lesions. His plaque index was 3 and localized staining was present on teeth 13, 23, 32 and 42 (Figure 5, Figure 6). These minor abnormalities could be attributed to the boy’s inability to hold a toothbrush due to the resorptive deformities of his hands and wrists. Feeding and occasional tooth brushing were performed by his uncle who was his primary care-giver.
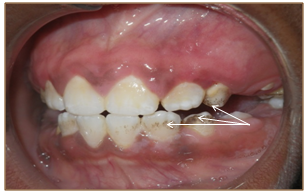
Figure 5: Stained teeth (arrows).
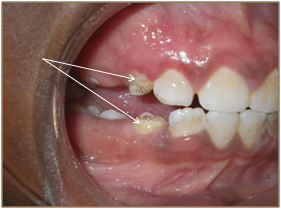
Figure 6: Visible plaque on the cervical region of his teeth (arrows).
The affected boy experienced difficulty in chewing due to tenderness in the TMJ area and a soft diet was necessary. This problem was most likely related to his partially erupted permanent molars and premolars. Mild gingival hypertrophy was noted in the posterior regions of both maxillary and mandibular jaws.
The delayed eruption of his permanent teeth and the integrity of his TMJ were matters of concern. and a Cone beam computed tomographic (CBCT) investigation was undertaken.
The radiographer encountered difficulty in positioning of the patient since movement of his head and jaw resulted in discomfort. Due to an incomplete field of view, vertebral body evaluation was impossible. Mild generalized osteoporosis was evident and asymmetry of the craniofacial structures were apparent (Figure 7). The nasopharyngeal airway was patent. Other airway spaces such as ethmoid air cells and nasal cavity appeared patent, but the right maxillary sinus was partially opacified (Figure 7). The right mandibular condyle head was hypoplastic and the surface cortical bone on the left condylar head was thin and uneven (Figure 8).
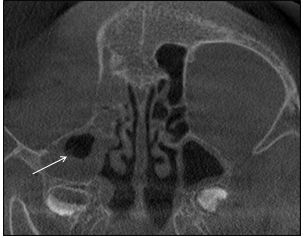
Figure 7: Coronal section of the craniofacial tissues. Generalized osteoporosis, asymmetry and partial opacification of the maxillary sinus (arrow) is evident.
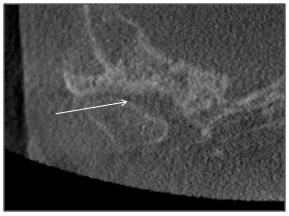
Figure 8: Coronal view of left mandibular condyle showing thin cortical bone and an uneven surface morphology (arrow).
Dental findings (Figure 9, Figure 10) Delayed development of the mandibular first molars with only two-thirds of root completion was evident. Normal permanent teeth eruption tables suggest that the first mandibular molars should have completely erupted by age 7 years with root completion by age 9.5 years[14,15]. There was also a delay in the root development of the first and second premolars. The coronal pulp chambers were large, suggestive of hypotaurodontism, and multiple intrapulpal calcifications were visible.
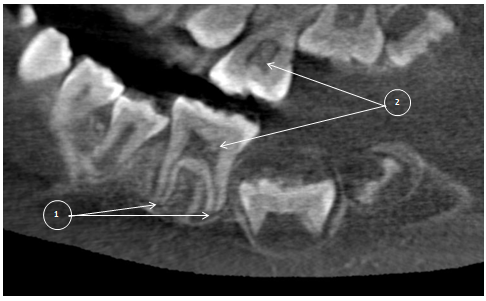
Figure 9: First mandibular molar (L) with 2/3rd of root development completed (1). Large coronal pulp chambers are suggestive of hypotaurodontism and contain several intrapulpal calcifications (2).
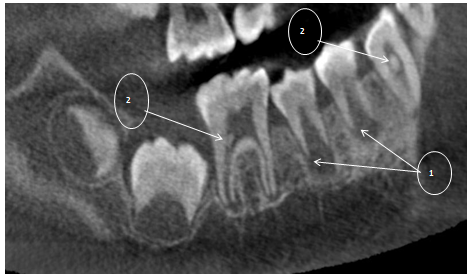
Figure 10: First mandibular molar (R) with 2/3rd of root development completed. Delayed development of the roots of the first and second premolars is depicted (1). Multiple intrapulpal calcifications are evident (2).
Comment
Skeletal lysis and consequent limb deformity is progressive in the osteolyses. Osteolysis of the carpal bones of both of the affected boy’s wrists had resulted in considerable impairment of the strength and control of his hands. He was unable to hold and use a toothbrush. Attempts at promoting independence have proved to be difficult and he is currently totally dependent on personal and social support from his uncle. The importance of dental hygiene was emphasized since routine dental care, such as, a scaling and polishing was very uncomfortable for the boy and challenging for the dental practioner.
Bisphosphonate treatment has been undertaken in view of the bone’s propensity to fracture following insignificant trauma. Nevertheless, the efficacy of bisphosphonate therapy in the osteolyses is uncertain as is the effect on the dentition[16].
The dental and craniofacial observations, presented in detail in this article, are primary and secondary manifestations of the disorder. They tend to be overshadowed by the more dramatic skeletal changes in the limbs. Nevertheless, appropriate dental care is an important component of the overall management of this severe condition.
Conflict of interest:
The authors declare no conflict of interest. All investigations were undertaken in complete accordance with the Declaration of Helsinki, the Hippocratic Oath and the Singapore Statement on Research Integrity. Formal ethical approval (HREC reference number: 203/2013) was obtained from the University of Cape Town’s ethics committee.
References
- 1. Bertie, J.D., Beighton, P., Thompson, D. The Torg-Winchester form of hereditary osteolysis: Orthopaedic manifestations and management. (2013) SA Orthop J 12(2): 23-27.
Pubmed || Crossref || Others - 2. Winchester, P., Grossman, H., Lim, W.N. et al. A new acid mucopolysaccharidosis with skeletal deformities simulating rheumatoid arthritis. (1969) Am J Roentgenol 106(1): 121-128.
Pubmed || Crossref || Others - 3. Torg, J.S., DiGeorge, A.M., Kirkpatrick, J.A. et al. Hereditary multicentric osteolysis with recessive transmission: A new syndrome. (1969) J Pediatr 75(2): 243-252.
Pubmed || Crossref || Others - 4. Zankl, A., Pachman, L., Poznanski, A. et al. Torg syndrome is caused by inactivating mutations in MMP2 and is allelic to NAO and Winchester Syndrome. (2007) J Bone and Min Res 22(2): 329-333.
Pubmed || Crossref || Others - 5. Prapanpoch, S., Jorgenson, R.J., Langlais R.P et al. Winchester syndrome. A case report and literature review. (1992) Oral Surg Oral Med Oral Pathol 74(5): 671-677.
Pubmed || Crossref || Others - 6. Zankl, A., Bonafe, L., Calcaterra, V. et al. Winchester syndrome caused by a homozygous mutation affecting the active site of matrix metalloproteinase 2. (2005) Clin Genet 67(3): 261-266.
Pubmed || Crossref || Others - 7. Beighton, P., Mennen, U., Golele, S.S. et al. Orthopaedic implications of heritable osteolysis in South Africa. (2007) SA Orthop J 6(2): 26-32.
Pubmed || Crossref || Others - 8. Al-Aqeel, A., Al-Sewairi, W., Edress, B. et al. Inherited multicentric osteolysis with arthritis: A variant resembling Torg syndrome in a Saudi family. (2000) Am J Med Genet 93(1): 11-18.
Pubmed || Crossref || Others - 9. Al.Mayouf, S.M., Majeed, M., Hugosson, C. et al. New form of idiopathic osteolysis: Nodulosis, arthropathy and osteolysis (NAO) syndrome. (2000) Am J Med Genet 93(1): 5-10.
Pubmed || Crossref || Others - 10. Al.Otaibi, L., Al.Mayouf, S.M., Al.Eid, W. et al. Radiological findings in NAO syndrome. (2002) Pediatr Radiol 32(7): 523-528.
Pubmed || Crossref || Others - 11. Rouzier, C., Vanatka, R., Bannwarth, S. et al. A novel homozygous MMP2 mutation in a family with Winchester syndrome. (2006) Clin Genet 69(3): 271-276.
Pubmed || Crossref || Others - 12. Vanatka, R., Rouzier, C., Lambert, J.C. et al. Winchester syndrome: the progression of radiological findings over a 23-year period. (2011) Skel Radiolog 40(3): 347-351.
Pubmed || Crossref || Others - 13. Bonafe, L., Cormier, V., Hall, C. et al. Nosology and Classification of Genetic Skeletal Disorders: 2015 Revision. (2015) Am J Med Genet 167A (12): 2869-2892.
Pubmed || Crossref || Others - 14. Al.Qahtani, S.J., Hector, M.P., Liversidge, H.M. Brief Communication: The London Atlas of Human Tooth Development and Eruption. (2010) Am J Phys Anthropol 142(3):481-490.
Pubmed || Crossref || Others - 15. Phillips, V.M., van Wyk Kotze, T.J. Dental Age Related Tables for Children of Various Ethnic Groups in South Africa. (2009) J Forensic Odontostomatol 27(2): 29-44.
Pubmed || Crossref || Others - 16. Bachrach, L.K., Ward, L.M. Clinical Review: Bisphosphonate use in childhood osteoporosis. (2009) J Clin Endocrinol Metab 94(2): 400-409.
Pubmed || Crossref || Others










