
The Role of the RAS Signaling Pathway in the Pathophysiology of Chronic Myelomonocytic Leukemia – New Insights from Molecular and Biologic Characterization
Affiliation
Fifth Medical Department with Hematology, Oncology and Palliative Medicine, Hospital Hietzing, Vienna, Austria
Corresponding Author
Prof. Dr. Klaus Geissler, MD, Fifth Medical Department with Hematology, Oncology and Palliative Medicine, Hospital Hietzing, Wolkersbergestraße 1, A-1130 Vienna, Austria, E-mail: klaus.geissler@wienkav.at
Citation
Geissler, K., et al. The Role of the RAS Signaling Pathway in the Pathophysiology of Chronic Myelomonocytic Leukemia - New Insights from Molecular and Biologic Characterization. (2017) Int J Hematol Ther 3(1): 1- 10.
Copy rights
© 2017 Geissler, K. This is an Open access article distributed under the terms of Creative Commons Attribution 4.0 International License.
Keywords
Chronic myelomonocytic leukemia; Pathogenesis; CFU-GM; RAS
Abstract
Recent insights into the pathophysiology of chronic myelomonocytic leukemia (CMML) have been obtained by the molecular and biologic characterization of primary leukemic cells from patients and from animal models. Almost 3 decades ago extensive myeloid colony growth in semisolid cultures without exogenous growth factors was observed as an in vitro characteristic of a subgroup of CMML patients. Recent data suggest that this phenomenon was probably the first indication of a hyperactive RAS signaling pathway in these patients. Although the mutation landscape in CMML is heterogenous and molecular aberrations in other signaling components can be found in some patients, the RAS pathway seems to play the major pathophysiological role in the majority of CMML patients with myeloproliferation (MP), disease progression and transformation into secondary acute myeloid leukemia (AML). There is also increasing evidence indicating that MP-CMML as a RAS pathway driven disease evolves from age related clonal hematopoiesis.
Introduction
Chronic Myelomonocytic Leukemia (CMML) is a hematopoietic malignancy of the elderly that is characterized by leukocytosis with monocytes and granulocytic cells in all stages of development, marked dysmyelopoiesis, a variable course, unresponsiveness to aggressive chemotherapy and an inherent risk of transformation to Acute Myeloid Leukemia (AML)[1,2]. With regard to the presence of myeloproliferation CMML was originally subdivided into myeloproliferative disorder (MPD)-CMML (WBC count > 13 x 109/L) versus myelodysplastic syndrome (MDS)-CMML (WBC count < 13 x 109/L MDS-CMML) by the FAB criteria[3]. Since CMML is characterized by features of both a MDS and a MPN the World Health Organization (WHO) classification of 2008 assigned CMML to the mixed category MDS/MPN[1]. CMML is further subclassified into CMML-1 ( < 5% circulating blasts and < 10% bone marrow blasts) and CMML-2 (5 - 19% circulating blasts, 10 - 19% BM blasts), with approximate median survival of 38 and 24 months, respectively[4-6].
In a large series of 1832 patients captured in the international CMML database that merged CMML registries from 8 tertiary cancer centers across 3 different countries between July 1981 and June 2014 the median age at diagnosis was 70 (16 - 93 years), with a male predominance (67%)[7]. Most patients were evenly subcategorized as MPN-CMML (49.8%) versus MDS-CMML (50.2%). Splenomegaly was demonstrable in 25% of all cases[7]. The median overall survival of CMML patients is about 30 months, one third evolving to AML while the others die from the consequences of cytopenias or comorbidities. Allogeneic transplantation, which is the only curative therapy, is rarely feasible because of age and/or comorbidities. In patients ineligible for transplantation, intensive chemotherapy results in low response rates and short response duration[8]. Hydroxyurea is used to control myeloproliferation[9]. The cytidine analogues azacytidine (AZA) and decitabine (5-aza-2′-deoxycytidine) have demonstrated some efficacy in delaying disease course in advanced CMML and were approved for the treatment of CMML[10-14].
High spontaneous formation of myeloid colonies without exogenous growth factors is an in vitro characteristic of a subgroup of patients with CMML
We have originally shown that extensive formation of colony-forming units granulocyte/macrophage (CFU-GM) without the addition of exogenous growth factors is an in vitro characteristic of a subgroup of patients with CMML (Table 1)[15]. This observation has been reproduced by others[16,17] and seems to be an in vitro phenomenon which is typical for CMML since it can be regularly demonstrated in CMML but is not a common finding in other MPNs including CML. Moreover we have shown that spontaneous colony growth from CMML cells can be markedly reduced by addition of antigranulocyte/macrophage colony-stimulating factor (GM-CSF) antibodies, but not by addition of antibodies against G-CSF, IL-3, or IL-6 suggesting that GM-CSF signaling may play a major role in the pathophysiology of this disease (Figure 1)[18]. High spontaneous CFU-GM growth ( > 100/105 MNC) could be found in 40% of CMML patients in a small retrospective study[19]. Moreover, CMML patients with high spontaneous CFU-GM growth ( > 100/105 MNC) have a much worse prognosis than patients with low colony growth in this study indicating a clinical significance of our observation (Figure 2)[19].
Table 1: In vitro cultures from patients with CMML using the CFU-C assay. Mononuclear cells from patients and normal individuals were cultivated in semisolid cultures with or without colony-stimulating activity (CSA). Data show in both CMML patients massively increased myeloid colony (CFU-C) growth as compared to controls and also the formation of CFU-C without the addition of exogenous CSA (adapted from Geissler, K., et al, Leuk Res 1988[15]).
| Source | CFU-C/2.5 x 104 MNC | |
|---|---|---|
| With CSA | P1 1 BM MNC | 910 |
| 6 Controls BM MNC | 19.8 + 8.5 | |
| Pt 2 PB MNC | 23.0 | |
| 6 Controls | 0.36 + 0.15 | |
| Without CSA | P1 1 BM MNC | 815 |
| 6 Controls BM MNC | 0.0 + 0.0 | |
| Pt 2 PB MNC | 27.0 | |
| 6 Controls | 0.0 + 0.0 |
CMML, chronic myelomonocytic leukemia; CSA, colony stimulating activity; BM, bone marrow; PB, peripheral blood; MNC mononuclear cells
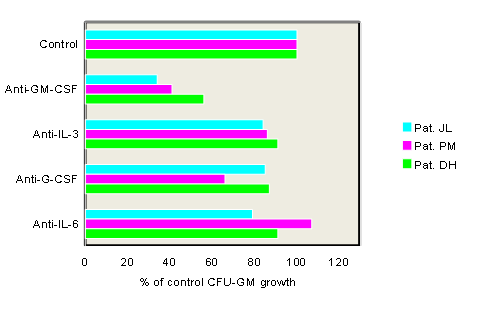
Figure 1: Effect of anticytokine antibodies on spontaneous growth of CMML cells in 3 patients. PB MNC were cultured with medium alone or with antibodies against GM-CSF, G-CSF, IL-3, or IL-6, respectively. Data show a marked inhibition of spontaneous CFU-GM growth in the presence of anti-GM-CSF antibodies in all 3 patients indicating that autonomous colony formation is a GM-CSF dependent in vitro phenomenon (adapted from Geissler, K., et al, J Exp Med 1996[18]).
CMML in preclinical models
The “RASopathies” are a group of genetic syndromes caused by germline mutations in genes that encode components of the RAS signaling pathway including NRAS, KRAS, NF-1, CBL, and PTPN11[20-25]. Besides their developmental defects they share a predisposition to juvenile myelomonocytic leukemia, a hematologic malignancy of early childhood. In the preclinical mouse model myelomonocytic leukemias can be recapitulated by transplantation of mouse BM cells harboring an oncogenic mutation in the Nras locus[26]. Interestingly, alterations of the other RASopathy genes may also lead to a similar phenotype in preclinical mouse models[27-31]. Mice develop a myeloprolifera tive disorder with clonal expansion of the granulomonopoiesis in vivo and show spontaneous in vitro myeloid colony formation without exogenous growth factors due to aberrant GM-CSF signaling.
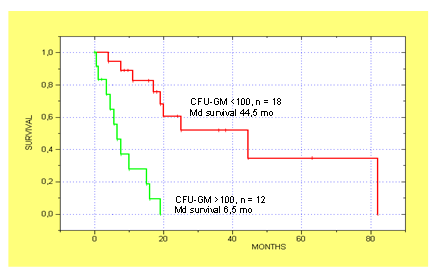
Figure 2: Kaplan-Meier estimates of survival for CMML patients with autonomous colony growth above (n = 12) and below (n = 18) 100 per 105 PBMNC. CMML patients with high CFU-GM growth had a significantly shorter survival than patients with low CFU-GM growth at presentation (median 6.5 vs. 44.5 months; p < 0.00002 by the log-rank test; adapted from Sagaster, V., et al, Ann Hematol 2004[19])
In an animal model conditional complete Tet2 loss leads to a progressive enlargement of the hematopoietic stem cell compartment and eventual myeloproliferation in vivo, including splenomegaly, monocytosis, and extramedullary hematopoiesis[32]. In addition, Tet (+/-) mice also displayed increased stem cell self-renewal and extramedullary hematopoiesis, suggesting that Tet2 haploinsufficiency contributes to hematopoietic transformation in vivo. Interestingly, another group could demonstrate that approximately 1/3 of Tet2(-/-) and 8% of Tet(+/-) mice died within 1 year of age because of the development of myeloid malignancies resembling characteristics of CMML suggesting that Tet2 loss may represent a predisposition for the development of this malignancy[33]. Furthermore it was shown by this authors that transplantation of Tet2(-/-), but not wild type or Tet2(+/-) BM cells, led to increased WBC counts, monocytosis and splenomegaly in wild type recipient mice[34].
Heterogenous mutational landscape in CMML
Thirty percent of CMML patients display an abnormal karyotype, with common abnormalities being +8 (23%), -Y (20%), -7/7q- (14%), 20q- (8%), +21 (8%) and der(3q) (8%)[34]. Molecular abnormalities are seen in > 90% of patients with CMML[35] with a marked genetic heterogeneity among CMML patients. Initial studies that characterized the mutational landscape in CMML focused on genes with a known myeloid leukemia or MDS association. Thus, a large number of gene mutations in genes encoding epigenetic regulators (TET2, ASXL1, EZH2, UTX, IDH1, IDH2, DNMT3A)[36-47], splicing factors (SF3B1, SRSF2, ZRSR2, 2AF1)[48-51], and signaling molecules (NRAS, KRAS, CBL, JAK2, FLT3)[52-58] have been found. Mutations in the transcription regulators RUNX1[58,59], NPM1[60], and TP53[61] have also been reported in CMML. Of all these mutations TET2 (~60%), SRSF2 (~50%), ASXL1 (~40%) and RAS pathway (~30%) are most common but no molecular aberration is specific of this entity, as they can be encountered with different frequencies in other myeloid neoplasms (Table 2)[62].
Table 2: Frequencies of recurrent genetic mutations in MDS/MPNs. Data show that of all mutations TET2, ZRSR2, ASXL1 and RAS pathway are most common in CMML but no molecular aberration is specific of this entity, as they can be encountered with different frequencies in other myeloid neoplasms (adapted from Padron, E., Hematology Am Soc Hematol Educ Program 2015[62]).
| CMML | JMML | aCML | MDS/MPN-U | RARS-T | |
|---|---|---|---|---|---|
| ASXL1 | 40 | 0 | 69 | ? | 15 |
| CALR | 0 | 0 | 0 | 0 | < 0 |
| CBL | 10 | 15 | 0 | ? | 4 |
| CSF3R | 0 | 0 | Variable | 0 | 0 |
| DNMT3A | 2 | 0 | ? | ? | 15 |
| ETV6 | < 1 | 0 | ? | ? | 3 |
| EZH2 | 5 | 0 | ? | 10 | ? |
| IDH1/2 | 6 | 0 | ? | ? | ? |
| JAK2 | 8 | 0 | 7 | 19 | 57 |
| JAK3 | NA | 12 | ? | ? | ? |
| K/N RAS | 19 | 39 | 35 | 14 | ? |
| NF1 | < 1 | 13 | ? | ? | ? |
| PTPN11 | < 1 | 44 | ? | ? | ? |
| RUNX1 | 15 | 0 | ? | 14 | ? |
| SETBP1 | 9 | 8 | 48 | 10 | 1 |
| SF3B1 | 6 | 0 | ? | ? | 98 |
| SRSF2 | 46 | 0 | ? | ? | 7 |
| TET2 | 58 | 0 | ? | ? | 25 |
| TP53 | < 1 | 0 | ? | ? | ? |
| U2AF1 | 5 | 0 | ? | ? | 5 |
| ZRSR2 | 8 | 0 | ? | ? | 3 |
Age related mutations in CMML
Recent genetic analyses of large populations have revealed that somatic mutations in hematopoietic cells leading to clonal expansion are commonly acquired during human aging[63]. Clonally restricted hematopiesis is associated with an increased risk of subsequent diagnosis of myeloid neoplasia. As some of the genes commonly mutated in age-related clonal hematopoesis such as TET2 and ASXL1 are also frequently mutated in CMML and aged hematopoiesis is characterized by a myelomonocytic differentiation bias it was recently hypothesized that CMML and aged hematopoiesis may be closely related[64]. By initially establishing the somatic mutation landscape of CMML by whole exome sequencing followed by gene-targeted validation it could be shown that most CMML patients (71%) had mutations in > 2 ARCH (age-related clonal hematopoiesis) genes and 52% had > 7 mutations overall. Higher mutation burden was associated with shorter survival. Age-adjusted population incidence and ARCH mutation rates suggests that CMML represents the leukemic conversion of the myelomonocytic-lineage-biased aged hematopoietic system.
There are several lines of evidence now that mutations in TET2 are in fact initial a clonal driver in CMML[65]. These include the high frequency of these mutations which can be detected in up to 60% of CMML patients[45,46,49], the fact that TET2 mutated clones can be detected in a small fraction of older subjects with clonal, but non-leukemic hematopoiesis[66-69], the competitive advantage of murine and human HSC invalidated for TET2[32,70,71] and the results of single-cell clonal tracking experiments indicating that a TET2 mutation, when present, is often the earliest recurrent genetic event in CMML[75].
High spontaneous myeloid colony growth in CMML reflects RAS pathway hyperactivation in patients with CMML
Molecular alterations of RASopathy genes in murine hematopoietic cells cannot only lead to a CMML-like disease in vivo but interestingly can also induce spontaneous myeloid colony formation in vitro due to hypersensitivity of granulomonocytic precursors against GM-CSF. Moreover, in JMML, in which molecular aberrations are mainly restricted to the RASopathy genes including NRAS, KRAS, NF1, CBL and PTPN11[73,74] the spontaneous formation of CFU-GM due to GM-CSF-specific hypersensitivity is a hallmark feature of disease[75], which has been included in the diagnostic criteria.
In a small preliminary study we recently observed a high prevalence of RAS pathway mutations in CMML patients with high colony growth (Figure 3)[76]. We therefore speculated that spontaneous myeloid colony formation might be a surrogate parameter of RAS pathway hyperactivation in CMML. To test this hypothesis we performed next-generation sequencing from stored peripheral blood mononuclear cells (PB MNC) obtained from 100 CMML patients, in whom in vitro cultures have been performed during the last years. In vitro culture data were then correlated with molecular aberrations of RAS pathway components[77]. In 40 CMML patients mutations in at least one of the RASopathy genes were detected, in 60 patients no mutations in RAS pathway components or such mutations with allele frequencies < 20% were found. In the 40 patients with RAS pathway mutations we found molecular aberrations of the NRAS gene in 19, KRAS in 6, NF1 in 3, CBL in 10 and PTPN11 in 2 patients, respectively. Mutations of RAS pathway components were mutually exclusive, only low levels of more than one RAS opathy mutation were found in some patients. In all patients with RAS pathway mutations additional mutations were observed in other genes, particularly in components of DNA methylation and/or the spliceosome as previously reported by othersA[36-51]. Results of semisolid cultures show that CMML patients in whom molecular aberration in RAS pathway components could be detected had a much higher spontaneous myeloid colony formation than CMML patients without RAS pathway mutations (Figure 4). The median number of spontaneously formed CFU-GM per 105 MNC was 147.5 (range 0 - 1009) in RAS positive patients as compared with 2 (0 - 812) in RAS negative patients (p < 0.00001 by the Wilcoxon’s rank-sum test). Unstimulated myeloid colony formation in RAS positive CMML patients was also much higher than the spontaneous formation of CFU-GM in normal individuals (median 4.8/105 PBMNC, range 3.5 - 8.5) which has been reported by us previously[78]. On the other hand the incidence of RAS pathway mutations was 72% (21/29) in CMML patients with high colony growth (>100/105 PBMNC) and 27% (19/71) in patients with low spontaneous CFU-GM formation (p < 0.0001 by the chi square test). In 8 patients high CFU-GM growth was observed without evidence of genetic aberrations in RAS signaling. This may indicate that additional molecular aberrations of the RAS pathway, that are not covered by our targeted NGS panel, may cause spontaneous cell proliferation, or alternatively, that other signaling pathways may also play a certain role in this in vitro phenomenon. There was no significant difference regarding autonomous CFU-GM growth in CMML patients with molecular aberrations in genes of epigenetic regulation and RNA-splicing, respectively. High spontaneous CFU-GM was never observed in CMML patients with the JAK2 V617F mutation as the only molecular aberration in signaling pathways (0/8 patients). Furthermore the in vitro conversion from a growth factor dependent to a growth factor independent phenotype by RAS but not by JAK2 could be demonstrated in BaF3 cells. Our findings suggest that high spontaneous myeloid colony growth in CMML is significantly associated with molecular aberrations of genes involved in RAS signaling and thus seems to reflect RAS pathway hyperactivation in patients with CMML.
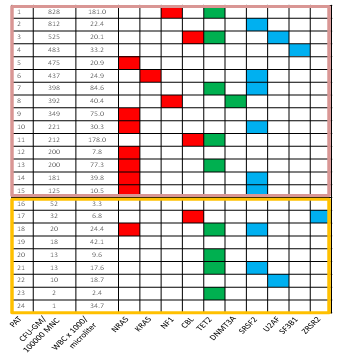
Figure 3: Mutation profiles in CMML according to spontaneous CFUGM growth. Each row corresponds to one patient. The first column indicates the patient number, the second the number of CFU-GM per 105 Peripheral Blood Mononuclear Cells (PBMNC), the third the White Blood Cell (WBC) count and all other columns represent the status of the genes. Colored squares indicate mutated, white squares wild-type genes. The colors of mutant genes indicate the most affected functional categories. Red, green, and blue indicate RAS pathway, DNA methylation, and spliceosome, respectively. Mutations in the components of the RAS pathway were found in 12/15 (80%) CMML patients with high colony growth ( > 100 CFU-GM/105 PBMNC) and in 2/9 (22%) patients with low spontaneous colony formation ( < 100 CFU-GM/105 PBMNC) (adapted from Geissler, K., et al, EHA Annual Meeting Abstracts 2015 (abstr E1300)[76]).
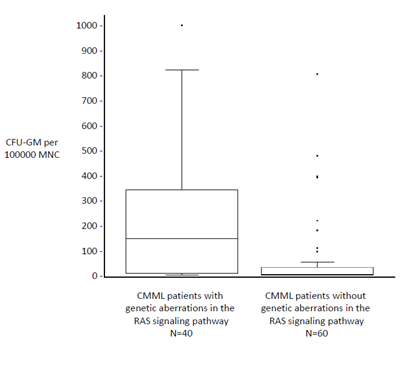
Figure 4: Comparison of spontaneous in vitro CFU-GM growth from PBMNC in CMML patients with and without mutations in RASopathy genes including NRAS, KRAS, NF1, CBL and PTPN11. CFU-GM formation in the absence of exogenous cytokines was assessed using semisolid cultures. Colony numbers are shown as box plots with first and third quartiles and interquartile ranges. Data were analyzed using the Wilcoxon’s rank-sum test. Spontaneous myeloid colony formation was significantly higher in CMML patients with mutations in RAS pathway components than in patients without such mutations (p < 0.00001). (adapted from Geissler, K., et al, Leukemia 2016, in press[77])
Correlation between high CFU-GM growth and myeloproliferation In CMML some patients present with only modest leukocytosis while others have high WBC counts and organ involvement, eg splenomegaly, serous effusions, and lymph node or skin infiltration[4]. Accordingly, an arbitrarily chosen leukocyte count has been used to distinguish between a dysplastic type (MDS-CMML, WBC count < 13 x 109/L) and a proliferative type (MPD-CMML; WBC count > 13 x 109/L). One important issue, therefore, is the question if genetic and/or biologic characteristics of leukemic cells can shape the phenotype of disease. In a small retrospective study with 30 CMML patients we were able to show that CMML patients with high spontaneous CFU-GM formation ( > 100/105 MNC) had higher WBC counts and a higher incidence of marked splenomegaly as compared with patients with low myeloid colony formation (Table 3)[19]. In another small study (n = 40) MP-CMML patients showed a higher frequency of RAS mutations by sequencing compared to MD-CMML[79].
Table 3: Relation of autonomous colony growth to clinicobiological features. In vitro cultures containing more than 100 colonies per 105 PBMNC were considered as high colony growth, whereas colony numbers below this cut-off level were called low growth. Median numbers (ranges) are given (adapted from Sagaster, V., et al, Ann Hematol 2004[19]).
| Feature | Low growth (n = 18) | High growth (n = 12) | P value |
|---|---|---|---|
| Age (years) | 73.5(60 - 85) | 68.5(53 - 88) | NS* |
| Sex (M/F) | 14/9 | 7/5 | NS° |
| Splenomegaly (> 5 cm below costal margin) | 1/18(6%) | 7/12(58%) | 0.005° |
| Hemoglobin (g/dL) | 10.7 (7.8 - 13.6) | 10.2 (8.3 - 13.2) | NS* |
| Platelets (x 109/L) | 136 (14 - 709) | 82 (13 - 262) | 0.042* |
| WBC (x 109/L) | 16,200 (3,220 - 25,170) | 36,480 (7,820 - 93,860) | 0.016* |
| Monocytes (%) | 23(5 - 57) | 22.5(10 - 74) | NS* |
| Blasts in PB (%) | 0(0 - 2) | 1.5 (0 - 4) | 0.002* |
| Serum LDH (U/L) | 244 (129 - 970) | 348 (120 - 738) | 0.044* |
| Lysozyme (μg/ml) | 65(18 - 366) | 62 (14 - 246) | NS* |
| Excess of blasts in BM (> 5%) | 1/18 (6%) | 6/12 (50%) | 0.017° |
NS not significant, WBC white blood cells, PB peripheral blood, BM bone marrow
*P of χ² test
°P of U- test (Mann- Whitney)
In the “Austrian Biodatabase for Chronic Myelomonocytic Leukemia” (ABCMML) we retrospectively and prospectively collect hematological, clinical, biologic and molecular information of patients with CMML from different centers in a real life setting. Using data from this database we were recently able to confirm our previous observations in a much larger patient cohort (n = 137). The diagnosis of CMML was made according to diagnostic criteria of the WHO classification of 2008. In this study we compared the amount of leukocytosis and splenomegaly between CMML patients with high colony growth (> 100/105 MNC) as compared to patients with low CFU-GM formation[80]. In agreement with our previous study high spontaneous myeloid colony formation was associated with increased WBC counts, increased blast cells, more pronounced splenomegaly and also inferior survival. Thus, these findings indicate that high spontaneous in vitro myeloid colony formation is not only associated with the presence of RAS pathway mutations[77], but also with leukocytosis, and splenomegaly. These results suggest that the myeloproliferative form of CMML with high spontaneous colony growth is a mainly RAS pathway driven malignancy.
Progression of CMML and transformation into AML
Little is known about factors contributing to the transformation of CMML into secondary AML. There are some data on the role of RAS mutations in the progression of MDS to AML[81-83] but the contribution of RAS pathway mutations in the transformation process in CMML is poorly investigated. In a study with 22 patients classified as MD-CMML and in 18 patients classified as MP-CMML it was shown that MP-CMML patients had a higher frequency of RAS mutations compared with MD-CMML. In two patients who progressed from MDS-CMML to MP-CMML, allele specific PCR unveiled low levels of the RAS mutations predominating in the myeloproliferative phase at the time of myelodysplastic disease, documenting for the first time the expansion of a RAS mutated clone in concomitance with CMML evolution[79].
We have previously reported in a small study that in patients with CMML spontaneous colony growth after transformation was significantly increased as compared to CFUGM growth before transformation (Figure 5)[19]. Since we have shown that high spontaneous myeloid colony growth in CMMLreflects RAS pathway hyperactivation in patients with CMML we hypothesized that molecular aberrations in the RASopathy genes may be responsible for transformation. Using data from the ABCMML we were recently able to address this question in a much larger number of patients by characterizing the clinical, molecular and biologic features of patients with CMML who developed AML[84]. Transformation into AML was defined as a blast cell percentage of at least 20% in the peripheral blood and/or bone marrow. Of the 355 CMML patients studied, we identified 47 who had already CMML derived AML at the time of inclusion (group A), 80 patients who later had documented transformation into AML (group B) and 228 patients, in whom no transformation was recorded (group C). CMML patients who developed AML (group A+B) had a significantly inferior survival, lower hemoglobin value, higher LDH, and higher spontaneous CFU-GM formation than patients who did not develop secondary AML. The incidence of RAS pathway mutations in group A+B was significantly higher than in group C (67% vs. 37%). The incidence of TET2 mutations in group A+B was not significantly different from the incidence in group C. There was also no significant difference in the incidence of mutations with regard to other epigenetic modulators and to components of the spliceosome. Our data confirm that patients with CMML who transform into AML, not unexpectedly, have an unfavorable prognosis. In the majority of them the transformation process seems to be associated with hyperactivation of the RAS pathway.
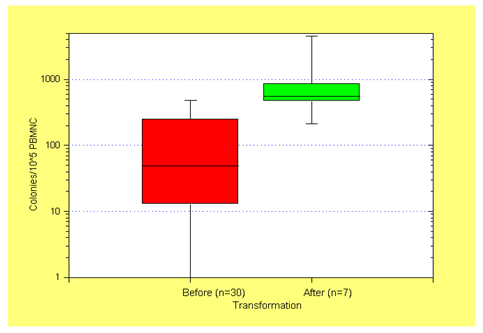
Figure 5: Autonomous CFU-GM growth in CMML patients before (n = 30) and after (n = 7) transformation into RAEBT/AML. The median values, the 25% – 75% percentiles, and the minimum and maximum values are given. Spontaneous colony growth after transformation into RAEBT/AML was significantly increased as compared to CFU-GM growth before transformation (p < 0.005 by the U-test; adapted from Sagaster, V., et al, Ann Hematol 2004[19])
RAS pathway mutations are uncommon in other myeloproliferative malignancies
Although RAS mutations may be sometimes observed in the transformation process of other hematologic malignancies molecular aberrations of components of the RAS signaling pathway are by far most prominent in JMML as it is known for a long time and in CMML as it has been shown by several groups including ours[76,85]. In one study NRAS mutations were not found in any of the 86 CML patients in blast crisis. Only one patient, in whom the initial diagnosis of CML blast crisis had been revised to CMML, displayed an NRAS mutation within codon 13[86]. In another study in which targeted cancer exome sequencing was performed in BCR-ABL-negative MPNs NRAS mutations were found in only 4.7% of 168 patients with primary myelofibrosis (MF) and no patient with Polycythemia Vera (PV)[87]. The transformation process in BCR-ABL-negative MPN has been also investigated by other investigators. Using single-nucleotide polymorphism microarrays to identify chromosomal aberrations, changes of chromosomes 1q, 7q, 5q, 6p, 7p, 19q, 22q and 3q were positively associated with post-MPN AML including commonly affected genes such as FOXP1, TET2, IKZF1, CUX1, ETV6 and RUNX1[88]. In another study somatic mutations with loss of heterozygosity in TP53 were strongly associated with leukemic transformation[89]. Although evolution of subclones of RAS pathway could be found in some patients, other molecular aberrations were more prevalent and significant. Moreover TET2 mutations seem to play an important role in the transformation process of BCR-ABL-negative MPN. Whereas data from us and others suggest that clonal disturbances of the epigenetic machinery usually precede the acquisition of RAS mutations and the emergence of a clone driven by hyperactivation of the RAS pathway in CMML, mutations in TET2 are commonly acquired at the time of leukemic transformation in BCR-ABL-negative MPN including MF, PV and essential thrombocythemia (ET)[90].
CMML with the JAK2 V617F mutation is a separate entity with hematological and biologic similarities to BCR-ABL-negative MPN
Whereas proliferative aspects in most CMML cases are related to aberrations in RAS signaling pathways a few cases have been reported to demonstrate JAK2 mutations[91-93]. The JAK2 V617F mutation which is a typical finding in PV and in around 50% of patients with ET and MF, respectively[94-98], is much rarer in CMML but can be consistently found in a subgroup of patients in larger series[6,45,49,92,99]. Due to the fact that JAK2 V617F-positive CMML is a rare disease the clinical, hematological and in vitro growth characteristics of this entity are poorly investigated. Recently, we characterized the clinical, hematological, molecular and biologic features of CMML patients with a JAK2 V617F mutation in our ABCMML[100]. We identified 13 CMML patients who had a JAK2 V617F mutation with an allele frequency > 20% who were compared to 105 patients who had NGS sequencing and were negative for the JAK2 V617F mutation. JAK2 V617F positive CMML patients had significantly higher WBC counts, higher hemoglobin values, higher platelet counts and more pronounced splenomegaly as compared to JAK2 V617F-negative patients. On the other hand the percentage on monocytes in peripheral blood and the number of myeloid colonies (CFU-GM) growing in vitro without addition of exogenous growth factors was lower in CMML patients with the JAK2 V617F mutation as compared to patients without this mutation. The majority of JAK2 V617F-positive patients had additional mutations that can be also found in JAK2 V617F-negative patients including mutations in genes encoding epigenetic regulators and splicing factors. Patients with the JAK2 V617F mutation had a trend towards a better OS than JAK2 V617F-negative patients. In a JAK2 V617F-positive CMML patient with splenomegaly, who was treated with the JAK1/2 inhibitor ruxolitinib off label, we could demonstrate the disappearance of constitutional symptoms and a durable spleen response lasting now for 56 months. Out data show that CMML patients with the JAK2 V617F mutation have hematological, biologic and clinical characteristics different from JAK2 V617F-negative CMML patients. These findings suggest that JAK2 V617F-positive CMML patients should be regarded as a distinct subgroup with hematological and biologic similarities to BCR-ABL-negative MPN which may benefit from specific pathogenesis oriented treatment concepts.
JMML and CMML with high spontaneous myeloid colony formation are both RAS pathway driven leukemias evolving from 2 distinct predisposition states
We believe that from a pathophysiological point of view there are significant similarities between JMML and MP-CMML. In both entities the RAS signaling pathway seems to play a major role in the expansion of the leukemic clone, the development of the phenotype and the progression of disease. This hypothesis is suggested by the fact, that in JMML and MP-CMML the incidence of mutations in components of the RAS signaling pathway is much higher than in any other hematologic malignancy[85] and that both entities show the typical spontaneous formation of CFU-GM in vitro which cannot be found in other entities to a similar extent[15,75]. Both entities seem to frequently develop on the basis of a genetic predisposition. JMML can be found in children with development disorders which are summarized by the term RASopathy. The RASopathies are a group of genetic syndromes caused by germline mutations in genes that encode components of the RAS signaling pathway including NRAS, KRAS, NF-1, PTPN11 and CBL[20,25]. Besides their developmental defects such as craniofacial dysmorphology, cardiac, cutaneous, and musculoskeletal abnormalities they share a predisposition to JMML. Molecular alterations of these components in murine hematopoietic cells may lead to a myelomonocytic leukemia like phenotype in vivo and to spontaneous myeloid colony formation in vitro due to hypersensitivity of granulocytic/monocytic precursors against GM-CSF. In contrast age related clonal hematopoiesis is likely to form an acquired predisposition state for the development of CMML in elderlypatients. In particular TET2 mutations are found in a high percentage of CMML patients on the one hand and are increasingly found in normal individuals beyond 50 who then have a significantly increased risk to develop myeloid blood cancer[68,69].
Although mutations in components of the RAS signaling pathway are highly prevalent in JMML and MP-CMML, differences between both leukemias exist with regard to the incidence of loss of function mutations of NF1. The absence of a functional tumor suppressor gene on one allele of the germline, as it is the case in children with NF, favors loss of function mutations to become functionally relevant, since only one additional somatic mutation in such a gene is sufficient to completely inactivate the inhibitory component of the RAS pathway which will then results in increased RAS signaling. In CMML, however, the predisposition associated with age related clonal hematopoiesis in elderly patients would favor additional mutations in general and one gain of function mutation in a stimulatory RAS pathway gene alone could become functionally relevant, sufficient to expand the malignant clone and to drive the phenotype. This hypothesis may easily explain the fact that loss of function mutations in NF1 are more prevalent in children with JMML than in CMML.
Due to the diversity of mutations found in CMML patients some investigators argue that CMML is a heterogeneous disease without common pathogenesis in contrast to JMML which is a more genetically homogenous disease and in which it is generally accepted that this leukemia is a RAS pathway driven malignancy[85]. However, in vitro findings by us[18] and others[101] suggest that divergent molecular aberrations in CMML seem to converge within the GM-CSF signaling pathway. Moreover, the higher number of mutations found in CMML might partly be explained by the fact that some of these mutations might be only clonal drivers that cause an acquired predisposition state for the development of additional mutations but actually have nomajor role in the myeloproliferation of the disease which eventuallyrequires treatment. Many of these mutations including TET2 can be found in normal individuals without overt leukemia suggesting that they are not necessarily contributing to the leukemic phenotype. In contrast mutations in genes of the RAS pathway components are usually not found in normal individuals and have been shown by us and others to be associated with progression of CMML and with transformation of CMML into secondary AML suggesting that mutations in these genes play a major role in expanding the malignant clone and causing the myeloproliferative phenotype. Therefore we think that there is sufficient evidence indicating that the myeloproliferative form of CMML is in the majority of patients a RAS pathway driven disease which is superimposed onto age related clonal hematopoiesis and that it could be beneficial to target this pathway by RAS pathway inhibitors to reverse the phenotype of the disease.
Author Contributions:
KG reviewed the literature and wrote the manuscript.
Conflict of interest:
The author declares no conflict of interest
References
- 1. Vardiman, J.W., Thiele, J., Arber, D.A., et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. (2009) Blood 114: 937-951.
Pubmed || Crossref - 2. Germing, U., Strupp, C., Knipp, S., et al. Chronic myelomonocytic leukemia in the light of the WHO proposals. (2007) Haematologica 974-977.
Pubmed || Crossref - 3. Bennett, J.M., Catovsky, D., Daniel, M.T., et al. Proposals for the classification of the myelodysplastic syndromes. (1982) Br J Haematol 51: 189-199.
Pubmed || Crossref - 4. Onida, F., Kantarjian, H.M., Smith, T.L., et al. Prognostic factors and scoring systems in chronic myelomonocytic leukemia: a retrospective analysis of 213 patients. (2002) Blood 99: 840-849.
Pubmed || Crossref - 5. Patnaik, M.M., Padron, E., LaBorde, R.R., et al. Mayo prognostic model for WHO-defined chronic myelomonocytic leukemia: ASXL1 and spliceosome component mutations and outcomes. (2013) Leukemia 27: 1504-1510.
Pubmed || Crossref - 6. Itzykson, R., Kosmider, O., Renneville, A., et al. Prognostic score including gene mutations in chronic myelomonocytic leukemia. (2013) J ClinOncol 31: 2428-2436.
Pubmed || Crossref - 7. Padron, E., Garcia-Manero, G., Patnaik, M.M., et al. An international data set for CMML validates prognostic scoring systems and demonstrates a need for novel prognostication strategies. (2015) Blood Cancer J 5: e333.
- 8. Padron, E., Steensma, D.P. Cutting the cord from myelodysplastic syndromes: chronic myelomonocytic leukemia-specific biology and management strategies. (2015) Curr Opin Hematol 22: 163-170.
Pubmed || Crossref - 9. Wattel, E., Guerci, A., Hecquet, B., et al. A randomized trial of hydroxyurea versus VP16 in adult chronic myelomonocytic leukemia: GroupeFrancais des Myelodysplasies and European CMML Group. (1996) Blood 88: 2480-2487.
Pubmed - 10. Fenaux, P., Mufti, G.J., Hellstrom-Lindberg, E., et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: A randomised, open-label, phase III study. (2009) Lancet Oncol 10: 223-232.
Pubmed || Crossref - 11. Braun, T., Itzykson, R., Renneville, A., et al. Molecular predictors of response to decitabine in advanced chronicmyelomonocytic leukemia: A phase 2 trial. (2011) Blood 118: 3824-3831.
Pubmed || Crossref - 12. Aribi, A., Borthakur, G., Ravandi, F., et al. Activity of decitabine, a hypomethylating agent, in chronic myelomonocytic leukemia. (2007) Cancer 109: 713-717.
Pubmed || Crossref - 13. Wijermans, P.W., Rüter, B., Baer, M.R, et al. Efficacy of decitabine in the treatment of patients with chronic myelomonocytic leukemia (CMML). (2008) Leuk Res32: 587-591.
Pubmed || Crossref - 14. Costa, R., Abdulhaq, H., Haq, B., et al. Activity of azacitidine in chronic myelomonocytic leukemia. (2011) Cancer 117: 2690-2696.
Pubmed || Crossref - 15. Geissler, K., Hinterberger, W., Bettelheim, P., et al. Colony growth characteristics in chronic myelomonocytic leukemia. (1988) Leuk Res 12: 373-377.
Pubmed || Crossref - 16. Everson, M.P., Brown, C.B., Lilly, M.B. Interleukin-6 and granulocyte-macrophage colony-stimulating factor are candidate growth factors for chronic myelomonocytic leukemia cells. (1989) Blood 74: 1472-1476.
Pubmed - 17. Ramshaw, H.S., Bardy, P.G., Lee, M.A., et al. Chronic myelomonocytic leukemia requires granulocyte-macrophage colony-stimulating factor for growth in vitro and in vivo. (2002) Exp Hematol 30:1124-1131.
Pubmed || Crossref - 18. Geissler, K., Ohler, L., Födinger, M., et al. Interleukin 10 inhibits growth and granulocyte/macrophage colony-stimulating factor production in chronic myelomonocytic leukemia cells.J ExpMed 1996; 184: 1377-1384.
Pubmed || Crossref - 19. Sagaster, V., Ohler, L., Berer, A., et al. High spontaneous colony growth in chronic myelomonocytic leukemia correlates with increased disease activity and is a novel prognostic factor for predicting short survival. (2004) Ann Hematol 83: 9-13.
Pubmed || Crossref - 20. Shannon, K.M., O'Connell, P., Martin, G.A., et al. Loss of the normal NF1 allele from the bone marrow of children with type 1 neurofibromatosis and malignant myeloid disorders. (1994)N Engl J Med 330: 597-601.
Pubmed || Crossref - 21. Kalra, R., Paderanga, D.C., Olson, K., et al. Genetic analysis is consistent with the hypothesis that NF1 limits myeloid cell growth through p21ras. (1994) Blood 84: 3435-3439.
Pubmed - 22. Tartaglia, M., Niemeyer, C.M., Fragale, A., et al. Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. (2003) Nat Genet 34: 148-150.
Pubmed || Crossref - 23. Loh, M.L., Vattikuti, S., Schubbert, S., et al. Mutations in PTPN11 implicate the SHP-2 phosphatase in leukemogenesis. (2004) Blood 103: 2325-2331.
Pubmed || Crossref - 24. Niemeyer, C.M., Kang, M.W., Shin, D.H., et al. Germline CBL mutations cause developmental abnormalities and predispose to juvenile myelomonocytic leukemia. (2010) Nat Genet 42: 794-800.
Pubmed || Crossref - 25. Niemeyer, C.M. RAS diseases in children. (2014) Haematologica 99: 1653-1662.
Pubmed || Crossref - 26. Wang, J., Liu, Y., Li, Z., et al. Endogenous oncogenic Nras mutation promotes aberrant GM-CSF signaling in granulocytic/monocytic precursors in a murine model of chronic myelomonocytic leukemia. (2010) Blood 116: 5991-6002.
Pubmed || Crossref - 27. Van Meter, M.E., Díaz-Flores, E., Archard, J.A., et al. K-RasG12D expression induces hyperproliferation and aberrant signaling in primary hematopoietic stem/progenitor cells. (2007)Blood 109: 3945-3952.
Pubmed || Crossref - 28. Li, Q., Haigis, K.M., McDaniel, A., et al. Hematopoiesis and leukemogenesis in mice expressing oncogenic NrasG12D from the endogenous locus. (2011) Blood 117: 2022-2032.
Pubmed || Crossref - 29. Parikh, C., Subrahmanyam, R., Ren, R. Oncogenic NRAS rapidly and efficiently induces CMML- and AML-like diseases in mice. (2006) Blood 108: 2349-2357.
Pubmed || Crossref - 30. Chan, R.J., Leedy, M.B., Munugalavadla, V., et al. Human somatic PTPN11 mutations induce hematopoietic-cell hypersensitivity to granulocyte-macrophage colony-stimulating factor. (2005) Blood 105: 3737-3742.
Pubmed || Crossref - 31. Le, D.T., Kong, N., Zhu, Y., et al. Somatic inactivation of Nf1 in hematopoietic cells results in a progressive myeloproliferative disorder. (2004) Blood 103: 4243-4250.
Pubmed || Crossref - 32. Moran-Crusio, K., Reavie, L., Shih, A., et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. (2011) Cancer Cell 20: 11-24.
Pubmed || Crossref - 33. Li, Z., Cai, X., Cai, C.L., et al. Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. (2011) Blood 118: 4509-4518.
Pubmed || Crossref - 34. Wassie, E.A., Itzykson, R., Lasho, T.L., et al. Molecular and prognostic correlates of cytogenetic abnormalities in chronic myelomonocytic leukemia: a Mayo Clinic-French Consortium Study. (2014) Am J Hematol 89: 1111-1115.
Pubmed || Crossref - 35. Patnaik, M.M., Tefferi, A. Cytogenetic and molecular abnormalities in chronic myelomonocytic leukemia. (2016) Blood Cancer J 6: e393.
Pubmed || Crossref - 36. Gelsi-Boyer, V., Trouplin, V., Adélaïde, J., et al. Mutations of polycomb-asso










