
Prolactin and Cisplatin Combination Treatment Inhibits Tumorsphere Formation and Delays Breast Tumor Growth in Mice
Eric H. Lee1, Andrew S. Mount1, Brian W. Booth2, Wen Y. Chen1*
Affiliation
- 1Department of Biological Sciences, Clemson University, Clemson, South Carolina
- 2Institute for Biological Interfaces of Engineering, Clemson University, Clemson, South Carolina
Corresponding Author
Wen Y.Chen, Department of Biological Sciences,Clemson University, 900 West Faris Road, Greenville, South Carolina 29605-4255. Phone: (864) 455-1457; E-mail: wenc@clemson.edu
Citation
Chen, W.Y., et al. Prolactin and cisplatin combination treatment inhibits tumorsphere formation and delays tumor growth in mice. (2015) Intl J Cancer Oncol 2(2): 1-7.
Copy rights
© 2015 Chen, W.Y. This is an Open access article distributed under the terms of Creative Commons Attribution 4.0 International License.
Keywords
Breast cancer stem cells; Prolactin; Cisplatin; Combination
Abstract
Cancer stem cells (CSC) are defined as a small population of cells in tumor that are responsible for the tumor initiation, resistance and recurrence. Chemo-resistance remains to be one of the major obstacles in conventional chemotherapies. One of the reasons that majority of chemotherapeutics are not effective in eradicating cancer cells is due to the existence of CSC, which are usually in a non-proliferative or dormant state. In this paper, we hypothesized that in order to improve the outcome of conventional chemotherapy, it will be more effective to utilize CSC stimulating factors in combination with conventional chemotherapeutics. The current study aimed to investigate the feasibility of using prolactin (PRL), a hormone intimately involved in mammary gland development, in combination with cisplatin as an alternative to target breast CSC (BCSC). Using tumorsphere formation assay we demonstrated that PRL was able to alter the cancer cell proliferation pattern in tumorspheres, which accompanied with increased CD44+24- cell population. We further showed that PRL treatment reduced the ability of breast cancer cells to form tumorspheres. Moreover, PRL significantly enhanced cisplatin's inhibitory effects in tumorsphere formation. The IC50 value of cisplatin was reduced by more than half with the addition of PRL in tumorsphere formation assay. The efficacy of this combinational approach was further confirmed through in vivo experiments (McNeu A allograft tumor growth and 4T1 total survival rate). Finally, PRL and cisplatin combination treatment significantly delayed naturally developed breast tumor growth in neu transgenic mice. Taken together, our study provided evidence to support the hypothesis that the addition of PRL to conventional chemotherapy (cisplatin) may be an effective alternative to target BCSC.
Introduction
There is increasing evidence suggests that cancer stem cells (CSCs) are a rare population of cells in tumors capable of self-renewal as well as differentiating into multiple lineages, thus responsible for tumor initiation, metastasis and recurrence[1]. Conventional cancer chemotherapy mainly targets proliferating cancer cells in a non-specific manner. Patients often experience adverse effects of chemo and radiation therapies throughout the course of treatment. Furthermore, failure of targeting CSC soften results in treatment resistance and cancer recurrence, which remain to be one of the major obstacles in conventional chemotherapy[2,3]. Tumors with high CSC markers are often associated with higher histological grade and poor prognosis[2,3]. Bonnet and Dick reported the existence of a cellular hierarchy in human acute myeloid leukemia (AML) by using stem cell markers CD34+/CD38-[4]. The isolated cells were able to transfer leukemia to immuno suppressed mice[4]. Breast CSCs (BCSCs) were first reported by Al-Hajj et al., and identified by surface marker expression of CD44+/CD24-/lin-[5] and can be even visualized with tumorsphere assay[6]. Despite the clinical efficacy of chemotherapeutic therapies, conventional treatments have not been shown to target BCSCs effectively[7,8]. Differentiation therapy has been proposed to be an alternative treatment approach in treating cancer[9,10]. As differentiated cells are more susceptible to chemotherapy[10], differentiation of BCSCs may increase susceptibility to chemotherapeutic agents. Previously Pece et al., demonstrated the similarities of transcriptional content between human normal mammary gland stem cells and cancer stem cells, in which poorly differentiated cancers displayed higher content of BCSCs than well-differentiated tumors[11].
In the current study we propose to use prolactin (PRL), a pituitary hormone responsible for mammary gland development and lactation[12-15], combined with chemotherapeutics to improve the chemotherapeutic outcome. The PRL hormone is critical for ductal cell differentiation into branches and terminal end alveoli cells[13]. During pregnancy, PRL promotes alveoli cell proliferate and lactation[15]. In breast cancer, high serum level of PRL has been documented to be associated with breast cancer[16]. PRL activates multiple signaling pathways including JAK/STAT[17], AKT[18] and MAPK[19] signaling pathways. Previous findings have also suggested that the addition of PRL to conventional chemotherapeutics prevents cells from cell cycle arrest and apoptosis[20]. Though PRL was thoroughly studied in previous reports, the role of PRL in BCSCs remains unknown in the field. In this study, we focus on the effect of PRL in combination with chemotherapeutics in a rare population of breast cancer cells, rather than the whole heterogenous cell population. Here we hypothesize that since PRL plays a critical role in regulating mammary gland proliferation and differentiation, PRL may also induce proliferation and differentiation of BCSCs, thus making them susceptible to those chemotherapeutic agents targeting proliferating and differentiated cells. By using the 3D tumorspheres culturing system, we were able to selectively study the effect of PRL in BCSC cells. The data presented in this study demonstrated that treatment with PRL decreased tumorsphere formation in a concentration dependent manner. Combination of PRL and cisplatin further reduced tumorsphere formation. Both mouse allograft model and neu mouse model demonstrated that PRL and cisplatin combination effectively delay tumor growth. Our data provides new insight into the effect of PRL in breast cancer.
Materials and Methods
Cell lines
Human breast cancer cell lines MCF-7 (ER+/PR+), HCC1954 (HER2+) and the murine mammary cancer cell line 4T1 were purchased from American Type Culture Collection (ATCC, Manassas, VA). The cells were propagated in RPMI supplemented with 10% FBS and 1 mg/mL Gentamicin (Life Technologies, Grand Island, NY). Mammary carcinoma cells from a neu transgenic mouse A (McNeu A, HER2+) were generously provided by Dr. Michael Campbell from the University of California, San Francisco. McNeu A cells were propagated in DMEM supplemented with 10% FBS and 1 μg/mL of Gentamicin. All three cell lines express high levels of PRLR based upon immuneblot experiments. Previous literature also demonstrated that MCF-7 cells[21] and McNeuA cells[22] express PRLR.
Tumorspheres culture
Tumorsphere medium was composed of DMEM/F12 (Sigma, St. Louis, MO) supplemented with 0.4% BSA, 5 μg/mL insulin (Sigma), 20 ng/mL basic fibroblast growth factor (Sigma) and 20 ng/mL epidermal growth factor (Sigma) unless otherwise specified[6]. Breast cancer cells MCF-7and HCC1954 were harvested at 80% confluency and seeded in ultra-low attachment flasks (Corning, Tewksbury, MA) at 2500 cells/mL. Cells were cultured in tumorsphere medium for 7 days before enzymatic dissociation for secondary tumorsphere formation in 24-well ultra-low attachment plates at 1000 cells per mL (Corning). Formation of secondary tumorspheres was evaluated after 7 days. For studies with PRL treatment, recombinant human PRL was produced and purified as described previously[23]. To study the effect of PRL in tumorspheres, cells were treated with PRL (500 ng/mL) for 7 days and tumorspheres were subsequently dissociated into single cells for secondary tumorsphere formation. These secondary tumorspheres received no treatment. Formation of secondary tumorspheres indicates the effects of PRL in primary tumorspheres in BCSCs. In contrast, experiments on PRL enhancing cytotoxic effect of cisplatin, PRL and cisplatin treatment were added in secondary tumorspheres culture and there was no treatment in primary spheres culture.
FACS analysis
Cells were washed three times in PBS prior to staining with CD44 antibodies conjugated with FITC and CD24 antibodies conjugated with PE. All antibodies for FACS were purchased from BD Biosciences (BD Biosciences, Franklin Lakes, NJ). Cells were incubated in 4°c for 30 minutes in dark and subsequently washed three times in PBS prior to FACS analysis.
PKH 26 assay
Breast cancer cells MCF-7 and HCC1954 were stained with PKH 26 fluorescence marker (PKH26GL-1KT; Sigma) according to manufacturer protocol prior to culture in tumorsphere medium. Briefly, cells were washed with PBS and stained with 2 μM of PKH 26 fluorescence marker in a non-specific manner for 10 minutes at room temperature. Subsequently cells were washed with PBS after staining and subjected to tumorspheres culture at 2500 cells per mL. Tumorspheres with 4-8 cells after 4 days of incubation were examined microscopically by Nikon Eclipse confocal microscope. Images are processed by NIS-elements. Validation of cell division pattern was performed in a double-blinded manner to avoid bias.
Immunoblotting
Total proteins were separated by 10% SDS-PAGE gel and subsequently transferred onto Hybond-ECL nitrocellulose membrane. Non-specific binding sites were blocked by TBST containing 5% non-fat powder milk for 1 hour and incubated overnight at 4°C with anti-pAKT antibodies (4060S, Cell Signaling, Danvers, MA) diluted at 1:1000, anti-AKT diluted at 1:1000 (sc-5928; Santa Cruz Biotechnology, Santa Cruz, CA), anti-pERK1/2 (sc-7383; Santa Cruz Biotechnology) diluted at 1:1000, anti-ERK1/2 diluted at 1:1000 (4695; Cell Signaling), or anti-β-actin (A1978; Sigma) diluted at 1:10000. Next, membranes were washed 3 times with TBST and incubated for 1 hour with HRP-conjugated goat anti-rabbit IgG or goat anti-mouse IgG antibodies (Bio-Rad, Hercules, CA) diluted at 1:2000. Western blot signals were detected by using ECL Western Blotting Substrate (GE Healthcare Sciences, Piscataway, NJ) and detected by CCD camera (Protein Simple, Santa Clara, CA).
Evaluation of the combination approach using three mouse medels
A. Allograft growth comparison of pre-treated McNeuA cells.
McNeuA cells were treated with cisplatin (2 μg/mL), PRL (100 ng/mL) and cisplatin (2 μg/ml), or untreated as control for 3 days in DMEM supplemented with 5% FBS. Treated cells were harvested and 5x105 cells were subsequently injected into mammary fat pads of neu transgenic mice. For best growth comparison, we injected cisplatin treated cells on the left side and PRL and cisplatin combination treated cells on the right side of the same mouse. Control tumor cells (cells with no treatment) and PRL treated cells were injected in separate mice in the same manner. Tumor growth was measured twice weekly, and tumor volume is calculated by (width2)*(length/2).
B. Inhibition of naturally developed breast tumor in neutransgenic mice
When tumor reached approximately 150 mm3 in diameter (usually at 6 months of age), mice were randomly divided into three treatment groups: (1), control (no treatment), (2) cisplatin, 5 mg/kg/week, i.p.; and (3) PRL (50 μg) daily and cisplatin (5 mg/kg/week) for 31 days. Tumor size was measured weekly, and tumor volume was calculated by (width²)*(length/2).
C. 4T1 BALB/c mice overall survival test.
Mouse mammary cancer 4T1 cells were injected (5x105 cells/mouse) into 12 week old BALB/c mice intravenously. Mice were randomly divided into three treatment groups, each group received intraperitoneal injections of (1) 5 mg/kg of cisplatin twice a week or (2) in combination with 50 μg PRL for the first week and 100 μg PRL for the remaining course of treatment, or (3) control. Survival of mice was observed daily.
Statistical Analyses
All statistical analyses were performed using GraphPad Prism version 5.01 for Windows (GraphPad Software, La Jolla, CA). Results are presented as the mean ± s.e.m. and were analyzed using Student t test unless otherwise specified. Statistical analyses for concentration response curve was based on the values of the top, bottom and EC50 or IC50. p < 0.05 was considered statistically significant.
Results
PRL induces symmetric division in BCSCs
We investigated the effect of PRL on BCSC division. The confocal microscopy analysis demonstrated that the proliferation pattern of tumorspheres in control or in cisplatin treatment resulted in minimal dispersal of PKH 26 fluorescence marker, indicating the initial cell division of BCSC is asymmetric (Figure 1A and C). The addition of PRL resulted in a more even distribution of fluorescence (Figure 1B and D) suggesting the BCSCs underwent symmetric division in response to PRL stimulation. The 3D intensity surface plots under each confocal image offered a different presentation with the same results. In order to avoid any bias on confocal images, we have conducted this particular experiment seven times and two times in a double-blinded manner. We also investigated if the above visual proliferation pattern alteration in tumorsphere induced by PRL was related to CD44+/CD24- population change. By using FACS analysis, we found that cells isolated from secondary tumorspheres after PRL treatment increased CD44+/CD24- population (Figure 1E). There is no CD44+/CD24- population difference between control and PRL treatment in 2D culture condition.
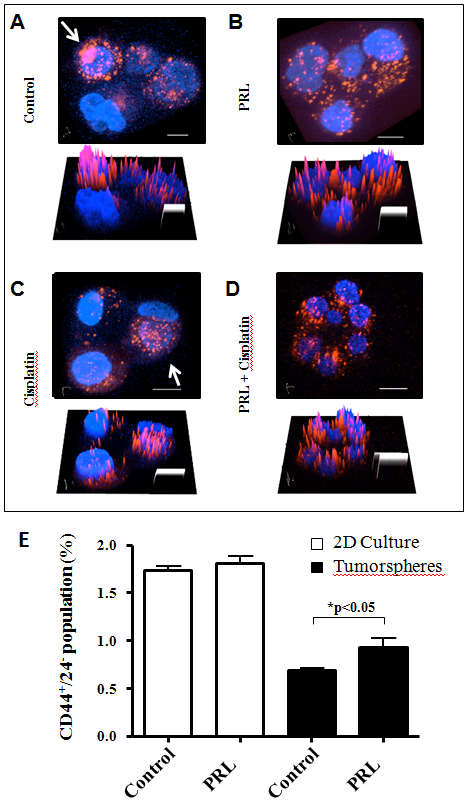
Figure 1: Confocal microscopic analysis of MCF-7 tumorspheres. MCF-7 cells were stained with PKH 26 fluorescence marker prior to tumorsphere culture with designed treatment conditions, A, control; B, PRL (500 ng/mL); C, cisplatin (2 μg/mL); and D combination of PRL and cisplatin. Arrows indicated cells retained high density of PKH 26 fluorescence marker in control or cisplatin groups suggesting the potential original stem cells. The bottom picture in each panel represents the fluorescence intensity in 3D surface plot. Blue, Hoechst 33342 nuclei fluorescence stain; Red, PKH 26 fluorescence marker. Original magnification: 400x. Scale bar, 20 μm. The result of a FACS analysis of CD44+/24- markers change in response to PRL in MCF-7 tumorspheres was shown in Panel E. *, p < 0.05.
PRL reduces tumorsphere forming ability in MCF-7 through PRLR
We next examined the effect of PRL in tumorspheres forming potential. In this experiment, we first collected primary tumorspheres formed in the presence or absence of PRL and dissociated the spheres into single cells for secondary tumorspheres formation without further exposure to PRL. We reasoned that the difference, if any, in the ability of formation secondary tumorspheres after PRL treatment indicates the BSCS amount in primary tumorspheres. We found that treatment with PRL in primary tumorspheres resulted in a 53% reduction of secondary tumorsphere formation from 64.3 ± 5.3 to 30.1 ± 4.3 per 1000 cells (p < 0.01) (Figure 2A). The inhibitory effect of PRL in total tumorsphere formation could be reversed with a PRL receptor antagonist, G129R, (Figure 2B), suggesting the PRL effect is PRL receptor (PRLR) specific.
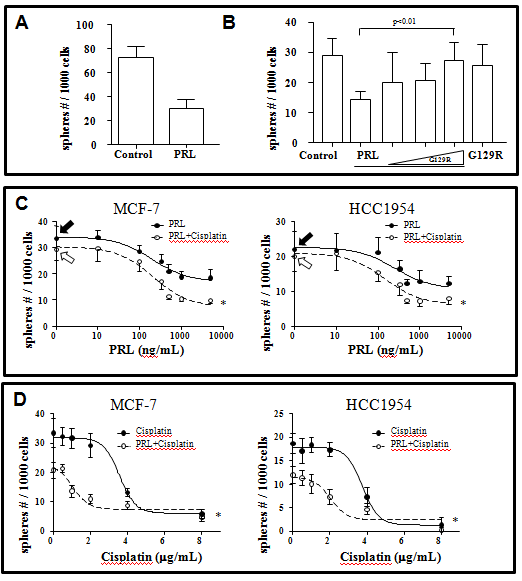
Figure 2: The effects of PRL on the secondary tumorsphere formation in MCF-7 and HCC1954 human breast cancer cell lines. There was a significant (53%) reduction of the tumorsphere formation in MCF-7 cells after PRL (500 ng/mL) treatment as compared to control (panel A). The inhibitory effect of PRL could be reversed by addition of increasing dose of a PRL antagonist, G129R (1 ug, 5 ug, and 10 ug, respectively, panel B). The concentration dependent response curves of PRL, with or without fixed dose of cisplatin (2 μg/mL), and the concentration dependent response curves of cisplatin, with or without fixed dose of PRL (500 ng/mL), were summarized in panels C and D. Close arrows represent control with no treatment at all. Open arrows represent cells treated with cisplain only. All data represented mean ± s.e.m. of five separate experiments.
PRL enhances cytotoxic effect of cisplatin in tumorsphere assay
We further tested if PRL will increase the efficacy of cisplatin through stimulation of BCSC using two breast cancer cell lines, MCF-7 and HCC1954. A PRL dose dependent inhibition of tumorsphere formation was first established in both MCF-7 (IC50 = 202 ng/mL) and HCC1954 cells (IC50 = 251 ng/mL) (Figure 2C). Maximum inhibitory effect of PRL is ~500 ng/mL in both MCF-7 and HCC1954 cells. Addition of a fixed dose of cisplatin (2 μg/mL) enhances the response of PRL, resulting lowered IC50 = 147 ng/mL in HCC1954 cells but not in MCF-7 cells (EC50 = 210 ng/mL). It is interesting to note that although there is no change in IC50, the maximum inhibition (total tumorspheres count) was enhanced (from 16.89 ± 2.73 to 7.21 ± 3.11) with the addition of cisplatin.
After obtaining the PRL response curve, we established complete dose-response curve for cisplatin in the presence or absence of the maximum dose of PRL in both cell lines. As presented in Figure 2D, PRL (500 ng/mL) significantly reduced the IC50 of cisplatin from 3.56 μg/mL to 1.03 μg/mL in MCF-7 cells and from 3.75 μg/mL to 2.05 μg/mL in HCC1954 cells.
PRL enhanced cisplatin treatment through the AKT and MAPK pathways
In order to test the effects of PRL and cisplatin combination, we compared tumor growth of allografts of McNeuA cells that were pre-treated in vitro with cisplatin or cisplatin plus PRL. Tumor outgrowth of this allograft experiment represents drug effects prior to transplantation. As expected there was a delay of McNeuA allograft tumor growth after cisplatin treatment when compared to control (Figure 3A). However, the PRL and cisplatin combination treatment was more effective in delaying tumor growth (Figure 3A). It is noticed that McNeuA cells treated with PRL had slightly increased tumor growth potential (no statistical difference). This observation should not be a concern since PRL was not proposed to be used alone. Immunoblotting analysis revealed that expression levels of pAKT and pERK were down-regulated by cisplatin. The inhibitory effect of cisplatin was enhanced with the addition of PRL (Figure 3B). In contrast, these two bio-markers behaved differently in tumors. There was a significant increase in pAKT and pERK in cisplatin pre-treated tumors (Figure 3C). It is important to point out that addition of PRL (combination treatment) would reverse the effect of cisplatin, i.e. reducing pAKT and pERK expression back to basal level.
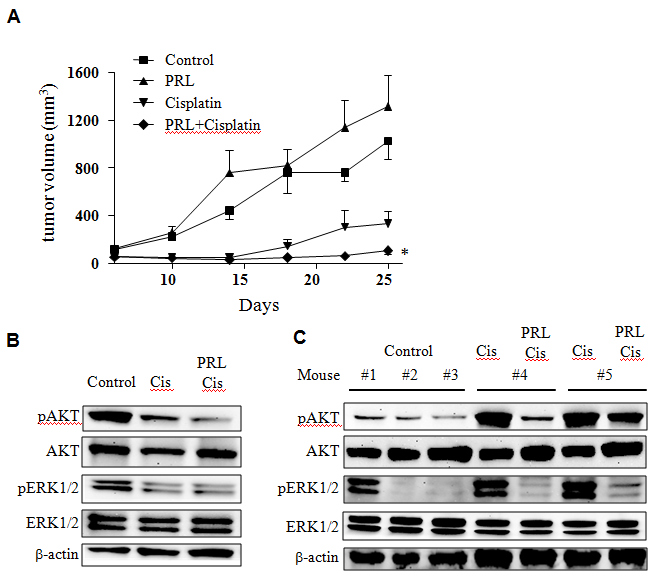
Figure 3: Comparison of MCneuA allograft tumor growth after cisplatin, PRL or cisplatin and PRL combination treatment. McNeu A cells that were pre-treated with cisplatin, PRL or combination in vitro for 3 days were harvested and inoculated into recipient female mice (n = 6/group). Tumor growth curves from each group were plotted for comparison (panel A). *, p < 0.05 as compared to cisplatin alone treatment. Immunoblotting of pAKT and pERK from McNeu A cells prior to implantation (panel B) or from allografted tumors removed at the end of the experiments (panel C) were performed.
PRL and cisplatin combination treatment delays tumor growth in naturally developed breast tumor in neu transgenic mice and prolongs overall survival in BALB/c mice
We examined the effectiveness of PRL and cisplatin combination treatment in naturally developed breast tumor (treatment starts when the tumor reached ~150 mm3 volume) using neu transgenic mice. Our results revealed that cisplatin alone treatment had little effect in inhibition of tumor growth whereas the combination of PRL and cisplatin significantly delays tumor growth (Figure 4A).We further tested combination efficacy using a more aggressive 4T1 mouse breast cancer metastatic model[24]. By tail vein injection of 4T1 cancer cells, we examined the survival time of the BALB/c mice. The median survival time of the cisplatin treatment group was significantly prolonged when compared to control (31 days vs 19 days). The addition of PRL to cisplatin treatment further extended the median survival time to 34.5 days (Figure 4B).
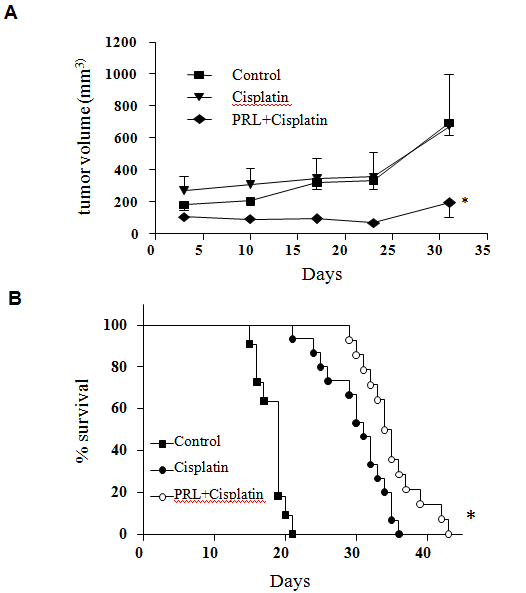
Figure 4: Comparison of naturally developed mammary tumors in neu transgenic mice in response to cispaltin or cisplatin and PRL combination treatment. Tumor bearing neu transgenic mice (tumors grew to 150 mm3 in diameter) were assigned randomly into three groups, control (n = 9); cisplatin (5 mg/kg, weekly, n = 9); or PRL (50 μg, daily) and cisplatin (5 mg/kg, weekly) combination (n = 10) for 31 days. Tumor growth curves from each group were plotted for comparison (panel A). *, p < 0.05 as compared to cisplatin alone treatment. Panel B was a Kaplan-Meier survival curve of cisplatin, or PRL and cisplatin combination treatment in 4T1 cell inoculated BALB/c mice. 4T1 cells were intravenously injected into BALB/c mice and randomized into three groups, control (n = 11); cisplatin (n = 15) and PRL and Cisplatin combination (n = 14). The combination treatment group resulted in the longest survival time of the three groups. Statistical significance was calculated by log-rank test. *p < 0.01 when compared to cisplatin treated group
Discussion
There is increasing evidence that breast cancer is driven and maintained by a small population of cells that exhibit stem cell properties[25]. Resistance of BCSCs to cytotoxic chemotherapy remains one of the major obstacles in successful cancer treatment[26,27]. In the current study we have demonstrated the principle of using a stem cell differentiation factor (PRL) in combination with a conventional chemotherapeutic (cisplatin) to improve the outcome of chemotherapy. By using tumorsphere and PKH 26 assays, we attempted to demonstrate that PRL modulates BCSC cell division patterns (Figure 1). Cicalese et al., reported that by staining cells with PKH 26 fluorescence marker prior to culture in tumorsphere-forming conditions, it is possible to identify the parental BCSC in a tumorsphere[28]. The data presented in Figure 1 and Supplementary 1 revealed that after exposure to PRL, BCSCs underwent symmetric division, evidenced by dispersed PKH 26 fluorescence marker in tumorspheres (Figure 1B and 1D). In contrast, minimal dispersal of the PKH 26 fluorescence marker is observed in the control or in the cisplatin treated groups (Figure 1A and 1C). To further confirm the effect of PRL on BCSCs, we compared the ability of tumorsphere formation from cells isolated from primary spheres. We were able to demonstrate that cells isolated from spheres after PRL treatment formed fewer tumorspheres in the subsequent passage (Figure 2A). These data indicate that PRL is able to modulate BCSCs division pattern, thus decrease the potential of tumorsphere formation. Such modulation of division pattern and decreased tumorspheres formation capacities indicate that PRL may have driven these cells into differentiation, since the treated cells exhibit differentiated cells characteristics, i.e. symmetrical division and losing tumorspheres formation abilities. These results provide supportive evidence to our hypothesis that PRL indeed stimulate CSC proliferation/differentiation.
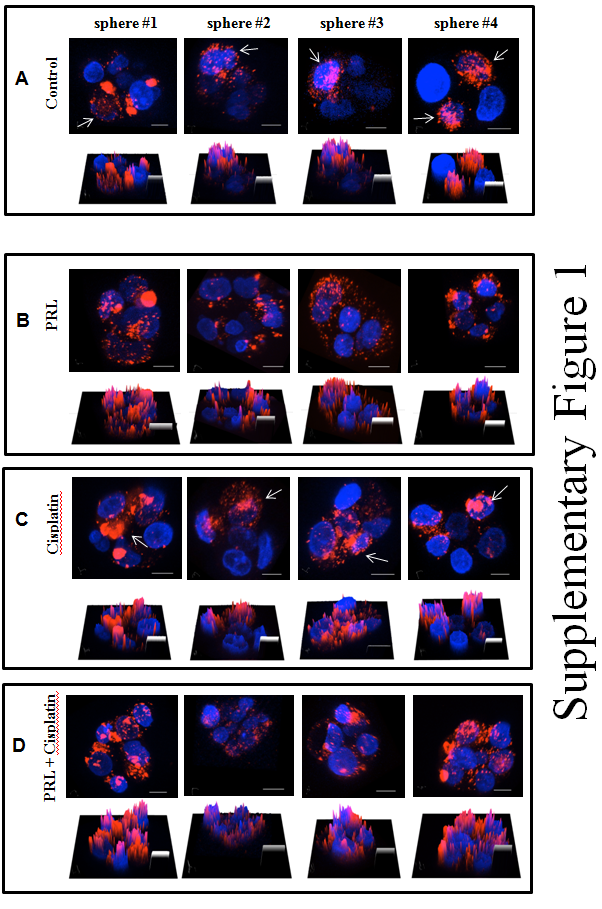
Supplementary figure: Confocal microscopic analysis of MCF-7 tumorspheresPanel A control. Panel B treatment of PRL (500 ng/mL). Panel C cisplatin (2 μg/mL). Panel D PRL and cisplatin combination. MCF-7 cells were stained with PKH 26 fluorescence marker prior to tumorspheres culture. Arrows indicated cells retained high density of PKH 26 fluorescence marker in control and cisplatin groups. Bottom pictures of each panel represent the fluorescence intensity in 3D surface plot. Blue, Hoechst 33342 nuclei fluorescence stain; Red, PKH 26 fluorescence marker. Original magnification: 400x. Scale bar, 20 μm.
Having established the cell division modulation effect of PRL on BCSCs (Figure 2A, 2B), we then investigated whether PRL could improve the effect of conventional chemotherapeutics. We were able to demonstrate that when cells were treated with cisplatin alone, the number of tumorspheres is marginally lower compared to control (Figure 2C), which agrees with previous findings that BCSCs are resistant to chemotherapeutics[27]. When cisplatin treatment was combined with PRL, the tumorsphere count was significantly decreased in a concentration dependent manner in both MCF-7 and HCC1954 cells (Figure 2C, 2D).
To further test the effects of PRL in BCSC, we pre-treated breast cancer cells with cisplatin or PRL and cisplatin combination and then engrafted these pre-treated cells into mice to observe if there was a difference in tumor cell growth potential. The reason that this modified proliferation assay was developed was because we could not detect any difference between cisplatin and PRL and cisplatin combination treatment by conventional cell proliferation assay. The effects of PRL (i.e. the combination treatment) could only be observed when pre-treated cells were engrafted into animals and the growth patterns were compared (Figure 3), which underscores the importance of an in vivo environment for the growth of tumor cells. Inactivation of Akt and MAPK was also observed from the harvested tumors, suggesting that the effects of PRL and cisplatin pre-treatment at molecular level was sustained and effective. The Akt and MAPK pathways have been reported to play central roles in chemoresistance[29,30]. Over expression of Akt and ERK is consistently observed in clinical samples that exhibit chemoresistance[30,31]. Our results suggest that PRL and cisplatin treatment reversed the Akt and ERK signaling cascade, which may be a contributing factor for the tumor growth delay.
In addition to the transplantation model, we also examined mammary tumor growth in response to PRL and cisplatin treatment using neu transgenic mice. Our results showed that continuous intra peritoneal administration of PRL and cisplatin significantly delayed tumor growth (Figure 4A). We further tested the PRL effect in BCSC by using 4T1 cells, a highly metastatic mouse mammary cancer model[24]. Our results showed that PRL and cisplatin combination treatment further increased the survival time as compared to cisplatin treatment alone (Figure 4B).
It is noteworthy to point out that a previous study by LaPensee et al., has demonstrated that treatment of breast cancer cells with PRL prevented cisplatin-induced G2/M cell cycle arrest and apoptosis[20]. It was also reported that in the presence of PRL, cisplatin was bounded to DNA, as determined by mass spectroscopy. The paper concluded that PRL confers resistance against cisplatin by activating a detoxification enzyme, thereby reducing drug entry into the nucleus. We believe that the difference in conclusion between our results and the results from LaPensee et al lies upon the cell culturing system. Studies in LaPensee et al utilized traditional 2D culturing system[20], in contrast, our in vitro study was performed in 3D tumorspheres condition. The major difference between the two culturing systems is that 3D tumorspheres culturing condition is specifically culturing proliferation and differentiation of stem/early progenitor cells, therefore, treatment of PRL is targeting a specific cell type rather than whole cell population in 2D monolayer. Recent studies have demonstrated that cells in 3D culturing conditions exhibited a different drug response than 2D monolayer condition[32-34]. Various culturing methods may contribute to different PRL response in breast cancer cells. It is also important to note that PRL and cisplatin combination treatment reduced tumor growth, tumorigenicity, down regulation of PI3K/AKT and MAPK pathways in vivo, suggesting the addition of PRL to cisplatin treatment deserves further investigation.
In summary, the data we presented showed that the addition of PRL to cisplatin treatment in breast cancer cells reduced their ability to form tumorspheres, suggesting that PRL may increase the susceptibility of BCSCs to cisplatin. We further demonstrated that PRL and cisplatin combination treatment delayed tumor growth and in mice. This study provides new insight that though PRL has stimulatory role in breast cancer, in principle, the addition of a differentiation factor to conventional chemotherapeutic treatment may be an alternative method targeting BCSCs.
References
- 1. Baccelli, I., Trumpp, A. The evolving concept of cancer and metastasis stem cells. (2012) J Cell Biol 198(3): 281- 293.
- 2. Siddique, H.R., Saleem, M. Role of BMI1, a stem cell factor, in cancer recurrence and chemoresistance: preclinical and clinical evidences. (2012) Stem Cells 30(3): 372- 378.
- 3. Ahmed, N., Abubaker, K., Findlay, J., et al. Epithelial mesenchymal transition and cancer stem cell-like phenotypes facilitate chemoresistance in recurrent ovarian cancer. (2010) Curr Cancer Drug Targets 10(3): 268- 278.
- 4. Bonnet, D., Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. (1997) Nat Med 3(7): 730- 737.
- 5. Al-Hajj, M., Wicha, M.S., Benito-Hernandez, A., et al. Prospective identification of tumorigenic breast cancer cells. (2003) Proc Natl Acad Sci U S A 100(7): 3983- 3988.
- 6. Dontu, G., Abdallah, W.M., Foley, J.M., et al. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. (2003) Genes Dev 17(10): 1253- 1270.
- 7. Tan, B., Piwnica-Worms, D., Ratner, L. Multidrug resistance transporters and modulation. (2000) Curr Opin Oncol 12(5): 450- 458.
- 8. Raguz, S., Yague, E. Resistance to chemotherapy: new treatments and novel insights into an old problem. (2008) Br J Cancer 99(3): 387- 391.
- 9. Leszczyniecka, M., Roberts, T., Dent, P., et al. Differentiation therapy of human cancer: basic science and clinical applications. (2001) Pharmacol Ther 90(2-3): 105- 156.
- 10. Azzi, S., Bruno, S., Giron-Michel, J., et al. Differentiation therapy: targeting human renal cancer stem cells with interleukin 15. (2011) J Natl Cancer Inst 103(24): 1884-1898.
- 11. Pece, S., Tosoni, D., Confalonieri, S., et al. Biological and molecular heterogeneity of breast cancers correlates with their cancer stem cell content. (2010) Cell 140(1): 62- 73.
- 12. Hennighausen, L., Robinson, G.W. Signaling pathways in mammary gland development. (2001) Dev Cell 1(4): 467- 475.
- 13. Hinck, L. Silberstein, G.B. Key stages in mammary gland development: the mammary end bud as a motile organ. (2005) Breast Cancer Res 7(6): 245- 251.
- 14. Hovey, R.C., Trott, J.F., Vonderhaar, B.K. Establishing a framework for the functional mammary gland: from endocrinology to morphology. (2002) J Mammary Gland Biol Neoplasia 7(1): 17- 38.
- 15. Brisken, C., O'Malley, B. Hormone action in the mammary gland. (2010) Cold Spring Harb Perspect Biol 2(12): a003178.
- 16. Eliassen, A.H., Tworoger, S.S., Hankinson, S.E. Reproductive factors and family history of breast cancer in relation to plasma prolactin levels in premenopausal and postmenopausal women. (2007) Int J Cancer 120(7): 1536- 1541.
- 17. Goffin, V., Bernichtein, S., Touraine, P., et al. Development and potential clinical uses of human prolactin receptor antagonists. (2005) Endocr Rev 26(3): 400- 422.
- 18. Chen, C.C., Stairs, D.B., Boxer, R.B., et al., Autocrine prolactin induced by the Pten-Akt pathway is required for lactation initiation and provides a direct link between the Akt and Stat5 pathways. (2012) Genes Dev 26(19): 2154- 2168.
- 19. Clevenger, C.V., Furth, P.A., Hankinson, S.E., et al. The role of prolactin in mammary carcinoma. (2003) Endocr Rev 24(1): 1- 27.
- 20. LaPensee, E.W., Schwemberger, S.J., LaPensee, C.R., et al. Prolactin confers resistance against cisplatin in breast cancer cells by activating glutathione-S-transferase. (2009) Carcinogenesis 30(8): 1298- 1304.
- 21. Scotti, M.L., Langenheim, J.F., Tomblyn, S., et al. Additive effects of a prolactin receptor antagonist, G129R, and herceptin on inhibition of HER2-overexpressing breast cancer cells. (2008) Breast Cancer Res Treat 111(2): 241- 50.
- 22. Xu, C., Langenheim, J.F., Chen, W.Y. Stromal-epithelial interactions modulate cross-talk between prolactin receptor and HER2/Neu in breast cancer. (2012) Breast Cancer Res Treat 134(1): 157- 169.
- 23. Langenheim, J.F., Tan, D., Walker, A.M., et al. Two wrongs can make a right: dimers of prolactin and growth hormone receptor antagonists behave as agonists. (2006) Mol Endocrinol 20(3): 661- 674.
- 24. Heppner, G.H., Miller, F.R., Shekhar, P.M. Nontransgenic models of breast cancer. (2000) Breast Cancer Res 2(5): 331- 334.
- 25. Kai, K., Arima, Y., Kamiya, T., et al. Breast cancer stem cells. (2010) Breast Cancer 17(2): 80- 85.
- 26. Abdullah, L.N., Chow, E.K. Mechanisms of chemoresistance in cancer stem cells. (2013) Clin Transl Med 2(1): 3.
- 27. Pinto, C.A., Widodo, E., Waltham, M., et al. Breast cancer stem cells and epithelial mesenchymal plasticity - Implications for chemoresistance. (2013) Cancer Lett 341(1): 56- 62.
- 28. Cicalese, A., Bonizzi, G., Pasi, C.E., et al. The tumor suppressor p53 regulates polarity of self-renewing divisions in mammary stem cells. (2009) 138(6): 1083- 1095.
- 29. Kim, D., Dan, H.C., Park, S., et al. AKT/PKB signaling mechanisms in cancer and chemoresistance. (2005) Front Biosci 10: 975- 987.
- 30. Chung, L.Y., Tang, S.J., Sun, G.H., et al. Galectin-1 promotes lung cancer progression and chemoresistance by upregulating p38 MAPK, ERK, and cyclooxygenase-2. (2012) Clin Cancer Res 18(15): 4037- 4047.
- 31. Oki, E., Baba, H., Tokunaga, E., et al. Akt phosphorylation associates with LOH of PTEN and leads to chemoresistance for gastric cancer. (2005) Int J Cancer 117(3): 376- 380.
- 32. Li, Q., Chow, A.B., Mattingly, R.R. Three-dimensional overlay culture models of human breast cancer reveal a critical sensitivity to mitogen-activated protein kinase kinase inhibitors. (2010) J Pharmacol Exp Ther 332(3): 821- 828.
- 33. Baker, B.M., Chen, C.S. Deconstructing the third dimension: how 3D culture microenvironments alter cellular cues. (2012) J Cell Sci 125(Pt 13): 3015- 3024.
- 34. Schyschka, L., Sánchez, J.J., Wang, Z., et al. Hepatic 3D cultures but not 2D cultures preserve specific transporter activity for acetaminophen-induced hepatotoxicity. (2013) Arch Toxicol 87(8): 1581- 1593.










