
Study on the Neuroprotection and Mechanism of C-EPO and EPO in MCAO Mice Models
Zhou Tong, Zhen-Ni Guo, Yu Dan Lv, Jiannan Zhu, Baoxin Ma, Qingqing Sun
Affiliation
The first hospital of Jilin University, 71 Xinmin Street, Changchun, Jilin, 130021, P. R. China
Corresponding Author
Zan Wang, Neurology Department, the first hospital of Jilin university, 71 Xinmin Street, Changchun, Jilin, 130021, P. R. China, Tel: +8643188782378, Fax: +8643188782378; E-mail: wangzanprof@163.com
Citation
Jian-hua, M., et al. Study on the Neuroprotection and Mechanism of C-EPO and EPO in MCAO Mice Models. (2017) Int J Hematol Ther 3(3): 1- 6.
Copy rights
© 2017 Jian-hua, M. This is an Open access article distributed under the terms of Creative Commons Attribution 4.0 International License.
Keywords
EPO; C-EPO; Stroke; Neuroprotection
Abstract
Background: Brain ischemia can be treated using two methods, namely thrombolysis and neuroprotection. The therapeutic time window for thrombolysis is less than 6 h, while that for most neuroprotective agents ranges from several hours to several weeks. Many stroke survivors can profit from neuroprotective treatment. Erythropoietin (EPO) is an interesting candidate neuroprotective agent in human patients with acute ischemic stroke, in addition to playing a role in red blood cell propagation. Carbamylated EPO (C-EPO) plays no role in red blood cell propagation, and its underlying mechanism of action in neuroprotection is unknown.
Objective: To understand the mechanisms underlying the neuroprotective effects of EPO and C-EPO in stroke.
Methods: Twenty one male mice with a Middle Cerebral Artery Occlusion (MCAO) were equally divided into 3 subgroups (n = 7 per subgroup): A1: MCAO; A2: 2500U/ kg EPO-MCAO; A3:C-EPO 25 μg/kg. MCAO models were developed by introducing a silicon-coated nylon monofilament into the common carotid artery and distally advancing it by 9 mm toward the carotid bifurcation. Ninety minutes following reperfusion, the behavior of the mice was observed. Finally, the mice from all the groups were decapitated, their brains were removed and sectioned, and the sections were stained with cresyl violet. Western blotting and immunohistochemistry were used to measure the expression of the IP3, ERK1/2, JNK, P38, JAK, STAT5, BCL-XL, and Caspase3.
Results: EPO and C-EPO increased the BCL-XL protein expression and decreased the Caspase3 protein expression in different manners. EPO can reduce apoptosis through activating IP3, MAPK, and JAK-STAT pathways, while C-EPO can reduce apoptosis only by activating the IP3 pathway.
Conclusion: EPO and C-EPO play a role in neuroprotection in the MCAO mouse model through similar mechanisms, possibly mediated by the modulation of the IP3 pathway.
Introduction
Many studies have shown that EPO plays an important role in neurotrophy, nerve growth, and neuroprotection. Kumral et al[1] found that the neuroprotective effect of EPO was better than that of IGF-1 or GDNF. The neuroprotective mechanism of EPO is more complex, and includes resisting free radical oxidation, glutamate generation, and apoptosis of nerve cells[2]. EPO and apoptosis have been extensively studied; EPO can activate Janus-tyrosine kinase 2 (JAK2), which is linked with the EPO receptor via its Box-1. In turn, JAK2 phosphorylates and activates downstream signaling factors, such as mitogen-activated protein kinase (MAPK)[3-5], extracellular-regulated kinase (ERK1/2), phosphatidylinositol 3 kinase (PI3K)/Akt, Jun kinase (JNK), and signal transducers and activators of transcription 5 (STAT-5). STAT-5 activates mitochondrial anti-apoptotic proteins, such as BCL-XL, and inhibits cytochrome C-dependent caspases (caspase3)[6,7]. EPO is routinely administered in patients with renal failure and anemia[8,9], and is considered safe in humans. However, its effects on the hematopoietic system limit its wide application in patients with stroke.
At present, there are two main EPO derivatives: Asialo-EPO and carbamyl-EPO (C-EPO). C-EPO has received much attention owing to its cytoprotective effect, and is known to leave the hematopoietic system unaffected. However, the signal transduction mechanism underlying the neuroprotective effects of C-EPO remains unclear. We hypothesized that the neuroprotective effect of C-EPO is related to JAK2-STAT, JAK2-MAPK, and JAK2-IP3 pathways. We investigated the effect of C-EPO and EPO on the expression levels of JAK2, STAT-5, ERK1/2, JNK, P38, Caspase3, and BCL-XL proteins in the Middle Cerebral Artery Occlusion (MCAO) mouse model, in order to understand the signal transduction mechanism underlying the neuroprotective effects of C-EPO and EPO. This will build the foundation for studying the clinical applications of C-EPO in ischemic stroke.
Methods
Experimental group
All the experimental procedures were approved by the first hospital of Jilin University for the Care of Experimental Animals. A total of 21 male C57BL/6 (weight, 20 - 25 g) mice, obtained from the Norman Bethune College of Medical Science Laboratories, Jilin university, were divided into 3 subgroups (n = 7 per group): A1: MCAO; A2: 2500U/kg EPO-MCAO; A3: C-EPO 25 μg/kg.
Carbamylation of EPO
EPO (ABCAM, USA) was carbamylated as described by Horkko et al[2]; EPO was incubated with 2 M cyanate for 6 h at 37°C, and the carbamylated EPO in each tube was used for dialysis. EPO was also carbamylated by incubation with 1 M cyanate for 6 h at 37°C. The carbamylated EPO (C-EPO) extensively dialyzed against 12 l of Phosphate-Buffered Saline (PBS) at pH 7.4 and a temperature of 4°C to remove excess cyanate. The C-EPO was used for the reaction with trinitrobenzenesulfonic acid after dialysis. The extent of carbamylation was monitored by following the loss of the free amino groups using trinitrobenzenesulfonic acid. Fifty microliters of 0.1 % trinitrobenzenesulfonic acid was added to 1 ml of EPO (500 units/ml in normal saline) and 1 ml of 4 % sodium hydrogen carbonate (pH 8.4), and incubated for 1 h at 37°C. The absorbance was then measured at 340 nm against a blank, and the trinitrobenzenesulfonic acid reactivity was expressed as a percentage of the absorbance obtained for the non-carbamylated EPO. The acylation rate of EPO was 85 %.
MCAO model generation
The mice were anesthetized with 1 % halothane (30 % O2; remainder, N2O). The rectal temperature was maintained at 37°C using a feedback-controlled heating system. The left common and external carotid arteries were isolated and ligated. A silicon-coated nylon monofilament was introduced into the common carotid artery and distally advanced by 9 mm toward the carotid bifurcation, thereby interrupting the MCA blood flow. Ninety minutes later, reperfusion was initiated by thread withdrawal. Immediately after reperfusion, 2 ml of 2500 U/kg EPO, or C-EPO 25 μg/kg was administered over a femoral venous catheter by continuous infusion over 30 min. The anesthesia was discontinued and the animals were placed back into their cages. Neurological and behavioral assessment was carried out 24 h later, and the mice were re-anesthetized and decapitated. The brains were removed, frozen on dry ice, and cut into 18-μ m-thick coronal sections on a cryostat. The brain sections were collected from five equidistant brain levels 2 mm apart, and were stained with cresyl violet. The tissue samples were obtained from both ipsilateral and contra lateral sides for the western blots. The stained brain sections were digitized, the brain infarcts were outlined, and the infarct volumes were measured.
Neurological and behavioral assessment
In order to detect sensorimotor impairments, an array of behavioral tests including the foot-fault and Modified Neurological Severity Score (mNSS) test were performed before and after the MCAO by an investigator blinded to the treatment group information.
Immunocytochemistry
The brain sections were washed in 0.1 M PBS thrice for 5 min each, and fixed in 4 % paraformaldehyde (in 0.1 M PBS) for 15 min. The sections were washed again in 0.1 M PBS thrice for 5 min each, and incubated in 0.1 M PBS containing normal goat serum (1:10 dilution) and 0.3 % Triton for 1 h. After three washes in 0.1 M PBS, the sections were incubated overnight at 41°C with polyclonal rabbit antibodies against JAK2 and STAT-5 (1:500 dilution). On the next day, the sections were washed in 0.1 M PBS thrice and then incubated for 1 h in anti-rabbit JAK2 and STAT-5 antibodies (1:250 dilution). After three washes, the sections were counter-stained with DAPI and cover slipped.
Western blotting
The tissue samples were incubated with lysis buffer, homogenized, and centrifuged. The supernatants were used for Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis (SDS-PAGE). The protein samples pooled from all the animals belonging to the same group were loaded onto the gel. Equal amounts of protein were diluted in 6X sample buffer, boiled, loaded on polyacrylamide gels, and transferred on to polyvinylidine fluoride membranes. Protein loading was controlled using a monoclonal mouse antibody against actin (SAB, USA). The membranes were incubated in blocking solution and immersed with monoclonal mouse β-actin, monoclonal mouse anti-diphospho ERK-1/-2(CST, USA), or polyclonal rabbit anti-diphospho JNK (CST, USA), P38 (CST, USA), IP3(SAB, USA), Akt (ABCAM, USA), BCL-XL (SANTACRUSE, USA), or Caspase 3(BD, USA) (1:500 dilution) in 0.1% Tween-20 (prepared in 0.1 M Tris-buffered saline). The membranes were washed, incubated with peroxidase-coupled secondary antibodies, washed, immersed in enhanced chemiluminescence (ECL) solution, and exposed to ECL-Hyperfilm. All the blots were replicated 3 times in order to confirm the reproducibility of the data. The protein levels were analyzed densitometrically and compared with values of samples derived from sham-treated control mice.
Statistics Analysis
The statistical differences between groups were calculated using one-way analysis of variance (ANOVA), followed by a post-hoc least significant differences test (SPSS for Windows 10.1). The values are presented as mean ± Standard Deviation (SD), with indicating the number of different samples analyzed. Results with P < 0.05 were considered statistically significant.
Results
Neurological deficits
Twenty-four hours after middle cerebral artery infarction, we evaluated the neurological impairment, and used the average standard deviation present in the neurological function score data. The neurological function score of each group was as follows: A1:2.3 ± 0.5; A2: 1.3 ± 0.9; A3: 1.5 ± 0.6 (Figure 1).
MCAO mouse models
Infarct volume
The infarction was observed in the right side of the brain cortex and striatum by using cresyl violet staining. The infarct volumes (mm³) of the A1, A2, and A3, groups were 56 ± 8.3, 28 ± 3.7, and 25 ± 3.2, respectively. The infarct volumes were significantly lower in the A2, and A3 groups than that in the A1 group (P < 0.05). There was no significant difference in the infarct volume among the A2 and A3 groups (P > 0.05) (Figure 1).
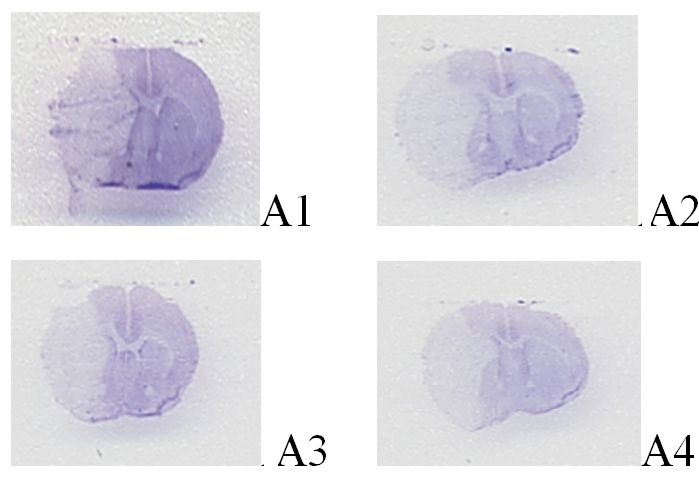
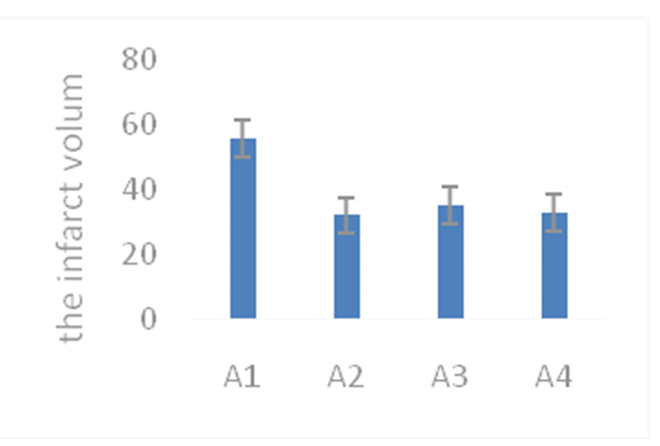
Figure 1: A) Cresyl violet staining of the 90-min middle cerebral artery occlusion (MCAO) mouse model. B) Infarct volume of the middle cerebral artery occlusion (MCAO) mouse model.
JAK-2 and STAT-5 expression in the MCAO mouse model (Figure 2, Figure 3)
The expression levels of JAK2 and STAT-5 in the different groups was studied using immunocytochemistry methods. The results showed that the expression level of JAK2 in the EPO and C-EPO groups significantly increased (Picture 2) (p < 0.05), compared with the expression level of JAK2 in the MCAO group. The expression level of STAT-5 in the EPO group increased (p < 0.05), whereas that in the C-EPO 25 μ g/kg MCAO groups did not increase , compared with the expression level of STAT-5 in the MCAO group (Figure 3) (p < 0.05).
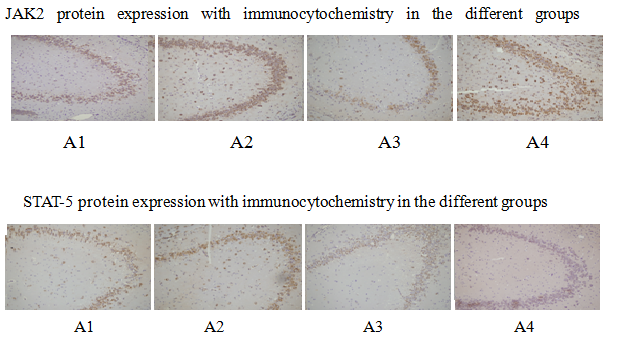
Figure 2: JAK2 protein expression with immunocytochemistry in the different groups; STAT-5 protein expression with immunocytochemistry in the different groups.
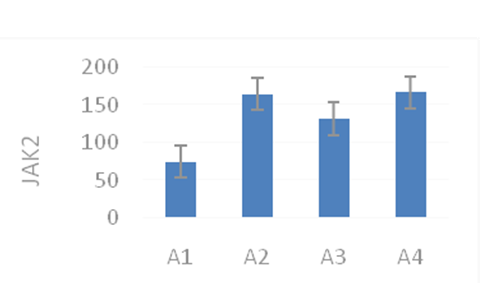
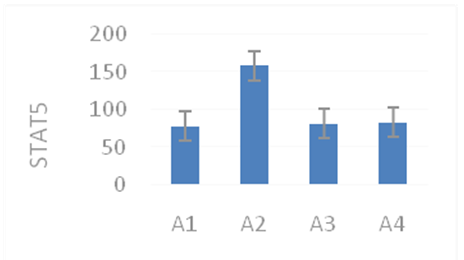
Figure 3: JAK2 and STAT-5 protein expression with immunocytochemistry in the different groups.
Actin, Erk1/2, Jnk, P38, IP3, AKT, BCL-XL, and Caspase3 expression levels in the different groups (Figure 4)
The expression levels of Actin, Erk1/2, Jnk, and P-38 in the different groups were studied using western blotting (Figure 4 A), which showed that the expression level of Jnk was higher in the EPO and C-EPO 25 μg/kg groups compared to that in the MCAO group. The expression levels of Erk1/2 and P-38 were not significantly different among the three groups.
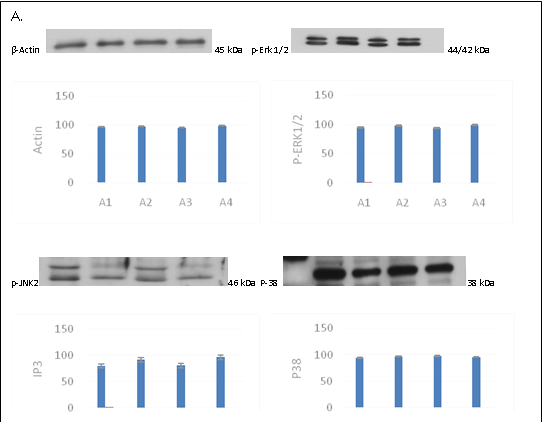
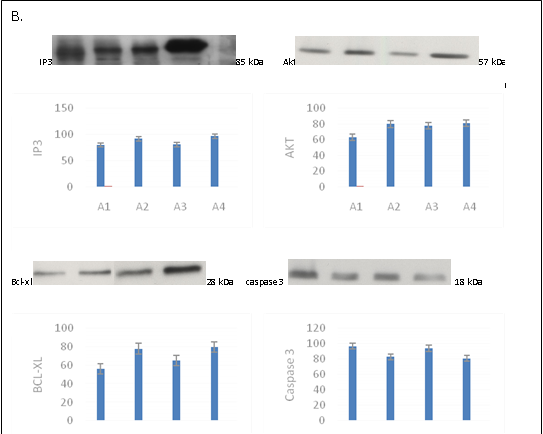
Figure 4: A. Actin, Erk1/2, Jnk, and P-38 protein expression in the different groups; B. The expression levels of IP3, BCL-XL, and Caspase3 in the different groups.
The expression levels of IP3, BCL-XL, and Caspase3 in the different groups was studied using western blotting (Figure 4 B), which showed that the expression levels of IP3 and BCLXL in the EPO and C-EPO 25 μg/kg groups were higher than those in the MCAO group. The expression level of Caspase3 was lower in the EPO and C-EPO 25 μg/kg groups compared to that in the MCAO group.
Discussion
In the present study, we found that EPO and C-EPO play a role in neuroprotection in the MCAO mouse models, but the mechanisms of neuroprotection is different, possibly mediated by the modulation of the IP3 pathway.
In 1948, Bonsdor and Jalsvisto[10] showed that erythropoietin (EPO) consists of 193 amino acids including sialic acid, which stimulates the proliferation and differentiation of red blood cells. EPO is synthesized and secreted by the tubules adjacent to cells of the renal cortex or medulla. Researchers have found that EPO is not only expressed in the liver and kidney, but also in the brains of rodents, monkeys, and humans[11,12]. In 1994, Masuda[13] et al found that the embryonic cells of the rat brain secreted EPO in vitro. There are two types of EPO receptors: EPOR and CD131. When EPO binds to the EPO receptor, it promotes the cross-linking of the EPO receptor to form a polymer, which is phosphorylated by a cytosolic tyrosine kinase, thereby activating the downstream intracellular signal transduction systems, including the JAK2-STAT-5 (through the activation of JAK1/2, STAT-3, and STAT-5), SOS-RAS-MAPK, and IRS-2-ph-K systems. In vitro studies have shown that EPO can activate JAK2, which plays an important role in EPO signal transduction, and trigger EPO signal transduction falls. It is a cytosolic protein connected to the BOX-1 cell receptor, which stimulates STAT-5 and activates mitochondrial anti-apoptotic protein BCL-XL after phosphorylation. Studies have shown that the anti-apoptotic effect of EPO disappeared in the JAK2 knockout model. Therefore, we should not rule out that JAK2 is necessary for the neuroprotective effect of EPO. Recent research shows that the neuroprotective effect of C-EPO can be independent of the EPO receptor. In this study, the expression level of STAT-5 was found to be higher in the EPO group than that in the MCAO group (p < 0.05), while the expression level of STAT-5 was not higher in the C-EPO 25 μg/kg MCAO groups, suggesting that the activation of STAT-5 did not participate in the neuroprotective effect of C-EPO. It is has been known that the MAPK pathway includes ERK1/2, JNK, and P-38; while the role of ERK1/2 in mediating the effect of EPO is not definite, YuKa Nagata et al[4] found that JNK and P-38 play a role in neuronal apoptosis Naetzker et al[14] also found that the apoptosis of neurons was related to the JNK and P-38 pathways. Our study showed that the expression level of JNK in the EPO and C-EPO 25 μg/kg groups was higher compared to that in the MCAO group A. However, there was no difference in the expression levels of ERK1/2 and P-38 among different groups, suggesting that the activation of JNK is important to the neuroprotective processes of EPO and C-EPO. The phosphatidylinositol 3-kinase (PI3K) pathway[15], the activation of phosphatidylinositol kinase (IP3K) can regulate the proliferation, differentiation, survival, and migration of cells. Our study showed that the expression level of BCL-XL was higher, while that of Caspase3 was lower in the EPO and C-EPO 25 μg/kg groups than that in the MCAO group. The expression level of IP3 was higher in the C-EPO 25 μg/kg group only than that in the MCAO group. Thus, EPO and C-EPO play the same role of neuroprotection in the MCAO mouse model, although the mechanisms underlying their effects are different. EPO and C-EPO reduce apoptosis, maybe through modulating the IP3 pathway. Advanced research[16] on developing methods to allow the EPO/C-EPO to cross the blood–brain barrier is necessary.
Acknowledge:
This study was supported by a grant from the Chinese National Natural Science Foundation, and the research grant of the Department of Science and Technology of the Jilin province (3D514S333428).
Conflict of interest:
The authors have no personal or financial conflicts of interest to disclose.
References
- 1. Kumral, A., Uysal, N., Tugyan, K., et al. Erythropoietin improves long-term spatial memory deficits and brain injury following neonatal hyoxia-ischemia in rats. (2004) Behav Brain Res 153(1): 77-86.
Pubmed || Crossref || Others - 2. Horkko, S., Savolainen, K., Kervinen, K., et al. Carbamylation-induced alternation in low-desity lipoprotein metabolism. (1992) Kidney Int 41(5): 1175-1181.
Pubmed || Crossref || Others - 3. Chin, K., Yu, X., Belesin-Cokic, B., et al. Production and processing of erythropoietin receptor transcripts in brain. (2000) Brain Res Mol Brain Res 81(1-2): 29-42
Pubmed || Crossref || Others - 4. Takahashi, N., Davis, R.J., Todokoro, K. Activation of p38 MAP Kinase and JNK But Not ERK Is Required for Erythropoietin-Induced Erythroid Differentiation. (1998) Blood 92(6): 1859-1869
Pubmed || Crossref || Others - 5. Ryan, J.J., Sawyer, S.T. JNK and p38 are activated by erythropoietin (EPO) but are not induced in apoptosis following EPO withdrawal in EPO dependent HCD57 cells. (2000) Blood 96(3): 933–940
Pubmed || Crossref || Others - 6. Lin, S.H., Kang, J.Q., Maiese, K. Erythropoietin prevents earlyand late neuronal demise through modulation of Akt1 and induction of caspase 1, 3, and 8. (2003) J Neurosci Res 71: 659–669
Pubmed || Crossref || Others - 7. Maiese, K. Apaf-1, Bcl-xL, cytochrome c, and caspase-9 form the critical elements for cerebral vascular protection by erythropoietin. (2003) J Cereb Blood Flow Metab 23(3): 320–330
Pubmed || Crossref || Others - 8. Sameri, R.M. Darbepoetin alfa, a new therapy for the management of anemia of chronic kidney disease. (2002) Pharmacotherapy 22(9 Pt 2): 141S-149S.
Pubmed || Crossref || Others - 9. Erythropoietin and renal failure. (2003) Curr Hematol Rep 2: 459-464.
Pubmed || Crossref || Others - 10. Jacobs, K., Shoemaker, C., Rudersdorf, R., et al. Isolation and characterization of genomic and cDNA clones of human erythropoietin. (1985) Nature 313(28): 806-810.
Pubmed || Crossref || Others - 11. Digicaylioglu, M., Bichet, S., Marti, H.H., et al. Localization of specific in defined areas of the mouse brain. (1995) Proc Natl Acad Sci U S A 92(9): 3717-3720.
Pubmed || Crossref || Others - 12. Marti, H.H., Wenger, R.H., Rivas, L.A., et al. Erythropoietin gene expression in human, monkey and murine brain. (1996) Eur J Neurosci 8(4): 666-676.
Pubmed || Crossref || Others - 13. Masuda, S., Okano, M., Yamagishi, K., et al. A novel site of erythropoietin production. Oxygen-dependent production in cultured rat astrocytes. (1994) J Biol Chem 269(30): 19488-19493.
Pubmed || Crossref || Others - 14. Naetzker, S1., Hagen, N., Echten-Deckert, G., et al. Activation of p38 mitogen-activated protein kinase and partial reactivation of the cell cycle by cis-4-methylsphingosine direct postmitotic neurons towards apoptosis. (2006) Genes Cells 11(3): 269-279.
Pubmed || Crossref || Others - 15. Montero, M., Poulsen, F.R., Noraberg, J., et al. Comparison of neuroprotective effects of erythropoietin (EPO) and carbamylerythropoietin (CEPO) against ischemia-like oxygen-glucose deprivation (OGD) and NMDA excitotoxicity in mouse hippocampal slice cultures. (2007) Exp Neurol 204(1): 106-117.
Pubmed || Crossref || Others - 16. Bulmer, C., Margaritis, A., Xenocostas, A. Encapsulation and controlled release of recombinant human erythropoietin from chitosan-carrageenan nanoparticles. (2012) Curr Drug Deliv 9(5): 527-537.
Pubmed || Crossref || Others










