
The Impact of the Novel ENAM Mutation on the Development of Hypoplastic Type of Amelogenesis Imperfect
Katarzyna Czarzasta1, Malgorzata Rydzanicz2, Sara Shamsa3, Malgorzata Zadurska4, Agnieszka Jurek4, Dorota Olczak-Kowalczyk3, Rafal Ploski2, Agnieszka Cudnoch-Jedrzejewska1
Affiliation
1Department of Experimental and Clinical Physiology, Laboratory of Centre for Preclinical Research, Medical University of Warsaw, Warsaw, Poland
2Department of Medical Genetics, Medical University of Warsaw, Warsaw, Poland
3Department of Paediatric Dentistry, Medical University of Warsaw, Warsaw, Poland
4Department of Orthodontics, Medical University of Warsaw, Warsaw, Poland
5Department of Histology and Embryology, Laboratory of Centre for Preclinical Research, Medical University of Warsaw, Warsaw, Poland
Corresponding Author
Pawel Wlodarski, Department of Histology and Embryology, Laboratory of Centre for Preclinical Research, Medical University of Warsaw, Banacha 1b, 02-097 Warsaw, Poland, Tel: (+48 22) 57 20 206; Fax: (0-22) 57 20 266; E-mail: pawel.wlodarski@wum.edu.pl
Citation
Wlodarski, P., et al. The Impact of the Novel ENAM Mutation on the Development of Hypoplastic Type of Amelogenesis Imperfect. (2018) J Dent Oral Care 4(1): 1- 4.
Copy rights
© 2018 Wlodarski, P. This is an Open access article distributed under the terms of Creative Commons Attribution 4.0 International License.
Keywords
Amelogenesis imperfect; ENAM gene; Enamelin; Mutations
Abstract
ENAM gene encodes ENAM elin, protein involved in mineralization of ENAMel. It has been previously demonstrated that certain mutations in ENAM gene lead to development of abnormal ENAMel, characteristic for the amelogenesis imperfecta (AI). In this case report we analysed the entire coding sequence of the ENAM gen in 6,5-years old girl diagnosed with ENAMel hypoplasia and suspected AI. Mutations were found in each allele of the ENAM gene p.Lys111Thr/c.332A > C and Leu420_Gly421fs/c.1260_1261insAG. One of these mutations p.Lys111Thr has not been described to date in association with ENAMel disease.
Introduction
Amelogenesis imperfecta (AI; MIM # 204700) is a group of developmental disorders of the ENAMel being transmitted as an autosomal dominant or autosomal recessive trait, or X-linked[1]. AI prevalence ranges from 1:2000 to 1:8000 in the world population[2]. The disorder is manifested by qualitative or quantitative ENAMel structure defects that may be accompanied by other dental and skeletal abnormalities such as tooth agenesis, delayed tooth eruption, coronal resorption, congenitally missing teeth, taurodontism, roots dilaceration, hyper cementosis, dental pulp calcification and malocclusion e.g. open bite[2].
AI is known to be caused by defects of six genes, including AMELX (amelogenin; (MIM * 300391)), ENAM (ENAMelin; (MIM *606585)), MMP20 (matrix metalloproteinase 20; (MIM *604629)), KLK4 (serine protease kalikrein 4; (MIM *603767)), FAM83H (casein kinase activity protein FAM83H; (MIM * 611927)), and WDR72 (WD repeat containing domain 72; (MIM *613214)), while the AMBN gene (ameloblastin; (MIM *601259)) is considered as a candidate gene[3]
Clinical manifestation of AI depends on the stage of ENAMel formation process that had been disturbed. There are four classes of AI: hypoplastic, hipomaturation, hypocalcified and hypomaturation with taurodontism[1]. The most characteristic clinical symptoms of AI are: extensively underdeveloped tooth tissues, increased tooth sensitivity, excessive tooth wear and spacing of anterior teeth[4].
The development of amelogenesis imperfecta has been associated with mutations of several genes, including ENAM[5,6]. This gene comprises of 10 exons, of which 8 are coding[7]. The mutations known to date were identified in seven exons and three introns[8]. The ENAM gene mutations in AI are inherited in an autosomal dominant (AI type IB) or autosomal recessive (AI type IC) pattern. ENAMelin is a critical matrix molecule in promoting crystal initiation and elongation during amelogenesis[9] and it plays an important role in mineralization of ENAMel.
ENAMelin is the largest, glycine-rich protein of the extracellular matrix of ENAMel (186 kDa). ENAMelin plays an important role in the mineralization of ENAMel, controlling growth of hydroxyapatite crystals in the ENAMel prisms. ENAMelin deficiency results in abnormal mineralization of the ENAMel, which therefore cannot withstand burden associated with the process of chewing[10].
ENAM gene mutations in amelogenesis imperfecta can cause variety of clinical symptoms ranging from generalized hypoplastic to pitted hypoplastic phenotype of AI[11]. Severity of syndromes, as well as co-existence of other dental and skeletal abnormalities can differ even in patients with the same mutation in the ENAM gene[12]. Therefore, further research on relationship between genotype and phenotype of these changes is needed. The aim of this study was to determine the clinical and genetic characteristics 6.5 - year-old patient diagnosed with AI.
Case Report
Clinical examination
The protocol of research and patient consent were verified and approved by the Department of Pediatric Dentistry and Department of Orthodontics at Medical University of Warsaw. Clinical examination of the 6.5-year-old patient and her parents consisted of physical examination and the record of the medical history. At the age of 7 years panoramic radiograph and at the age of 9 years cephalometric radiograph of the patient were taken. All clinical examination including blood sample collecting were conducted according to the Declaration of Helsinki.
Genetic study
In the patient DNA test was performed using the next generation sequencing method (NGS). The study was conducted at the Department of Medical Genetics, Medical University of Warsaw. The blood sample was taken from the patient for DNA isolation, then the clinical exome was sequenced (> 4800 genes) using TruSight OneTM Sequencing Panel Kit (Illumina, San Diego, CA, USA). Coverage of the sample in the NGS study was: 87.4% and 96.2%, for 20x and 10x minimal coverage, respectively. Obtained sequencing results were analyzed as previously described[13]. The occurrence of selected variants identified in the proband and considered as pathogenic was further confirmed by Sanger sequencing method and subsequently analyzed in patient’s healthy parents (DNA isolated from buccal mucosa).
Results
Clinical study
Patient was examined extraoraly, intraoraly and functionally and results of this examination were: (i) a slightly elongated lower facial height; (ii) mixed dentition, ENAMel hypoplasia of permanent incisors and first permanent molars, increased overjet and open bite (2 mm), teeth crowding, partial lack of space for permanent lateral incisors, especially in the mandible, low trailer frenulum of the upper lip and a short trailer tongue frenulum; (iii) hypotonia of the circular mouth muscle, incorrect rest position of the tongue, preserved infantile swallowing pattern, mouth breathing and impaired articulation.
Patient’s panoramic radiograph was taken at age of 7 years. All permanent tooth germs were present except of third molars, which may still be invisible at this age (Figure 1).
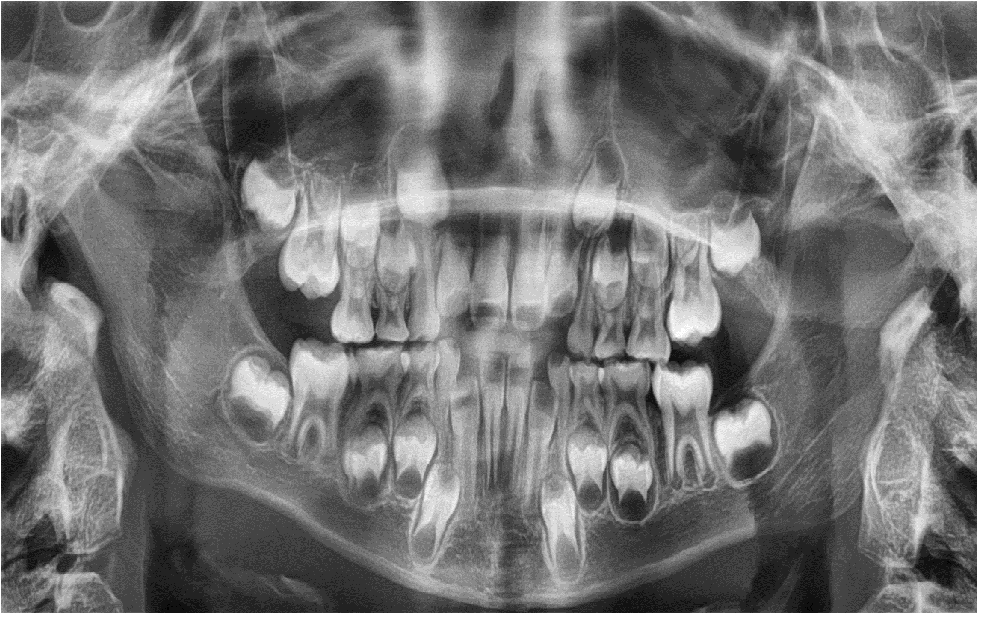
Figure 1: Panoramic radiograph at the age of 7 years.
Lateral cephalogram revealed a skeletal class II malocclusion and skeletal open bite (Table 1; Figure 2). In the course of treatment, the lower left lateral incisor had erupted ectopically with the loss of decidual canine. Dentition of both parents did not display any signs of developmental abnormalities.
Table 1: Selected cephalometric measurements according to Segner and Hasund and Steiner by Kaminek.
| Parameters | Norm | Result |
|---|---|---|
| SNA | 82.0 ± 3.0 | 76.6 |
| SNB | 80.0 ± 3.0 | 72.6 |
| ANB | 2.0 ± 2.0 | 4.1 |
| GntgoAr | 122.0 ± 7.0 | 132.8 |
| Index | 80.0 ± 7.0 | 79.2 |
| ML-NSL | 28.0 ± 5.0 | 44.4 |
| ML-NL | 20.0 ± 7.0 | 37.7 |
| NS-ML | 33.0 ± 6.0 | 46.5 |
| SGo:NMe | 60.5 ± 2.5 | 58.5 |
SNA- angle between sella, nasion and the A-point; SNB - angle between sella, nasion and the B-point; ANB - angle between the A-point, nasion and B-point; GntgoAr - angle between gnathion, gonion and articulate; ML-NSL - angle between the mandibular plane and the nasion-sella-plane; ML-NL - angle between mandible base plane and maxilla base plane, NS-ML - angle between cranial base plane and mandible base plane; SGo:NMe - percentage ratio between the posterior and anterior facial heights.
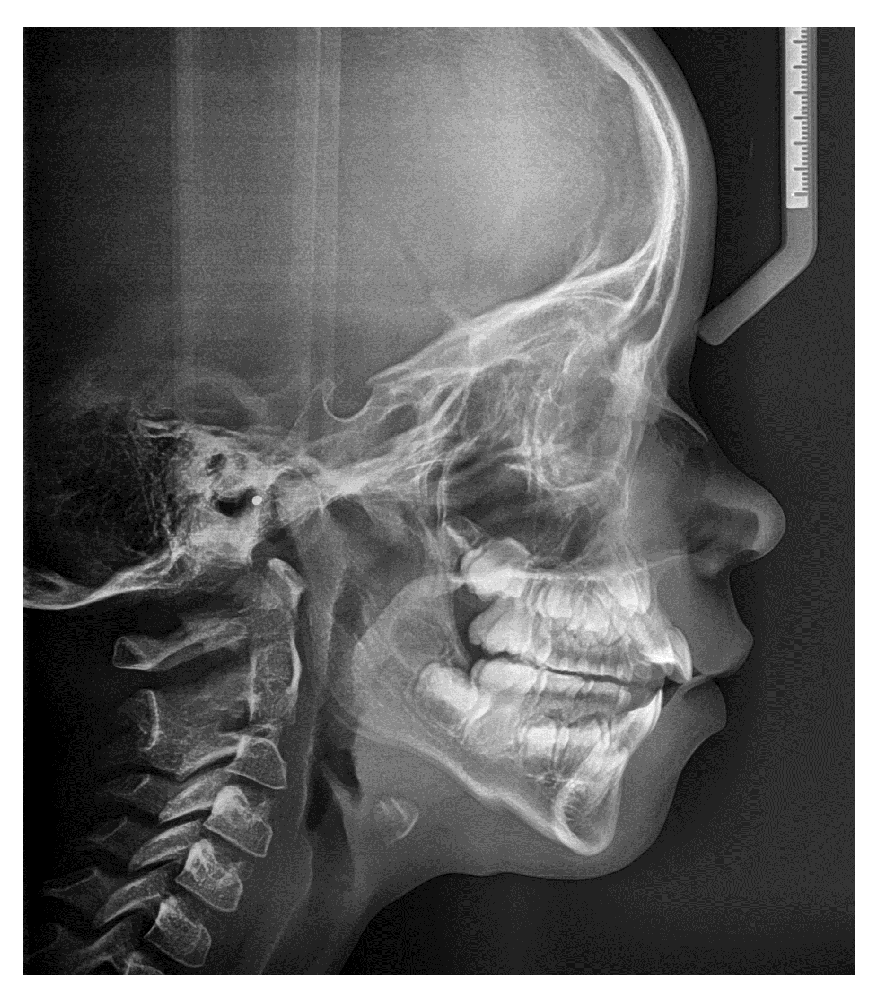
Figure 2: Lateral cephalogram made at the age of 9 years.
Genetic study
Sequencing of the clinical exome of the patient revealed two mutations in the ENAM gene (NM_031889.2): p.Lys111Thr/c.332A>C (rs532584416) and Leu420_Gly421fs/ c.1260_1261insAG (rs587776588). Both variants were confirmed in the proband by Sanger method (Figure 3). Additionally, the occurrence of these mutations was examined in parents of the proband. The Leu420_Gly421fs mutation was found in proband’s mother, while the p.Lys111Thr - in her father (Figure 4). The Leu420_Gly421fs mutation resulted in an insertion of two nucleotides (AG) between nucleotides 1259 and 1260 in the cDNA which causes a frameshift during translation resulting in addition of 26 essentially random amino acids and introduction of a premature termination at codon 448. To date, the ENAM p.Lys111Thr mutation has not been described in the literature in association with AI (https://www.ncbi.nlm.nih.gov/clinvar/). This mutation modifies the cDNA sequence at position 332 changing adenine (A) to cytosine (C), as a result in the ENAM protein lysine (Lys) at position 111 is replaced with threonine (Thr).
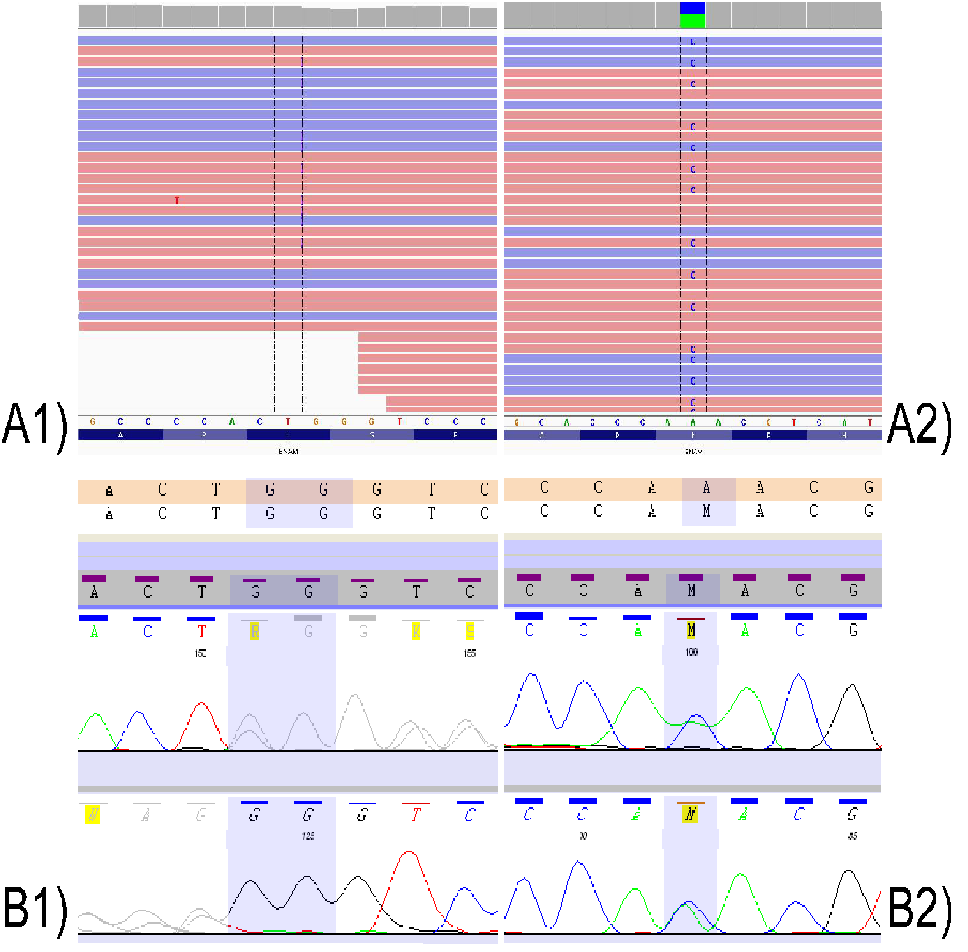
Figure 3: Mutational analysis of ENAM. A1) p.Leu420_Gly421fs/c.1260_1261insAG - sequencing of the clinical exome of the patient; A2) p.Lys111Thr/c.332A>C - sequencing of the clinical exome of the patient; B1) p.Leu420_Gly421fs/c.1260_1261insAG - mutational analysis in the proband by Sanger method; B2) p.Lys111Thr/c.332A>C - mutational analysis in the proband by Sanger methodd
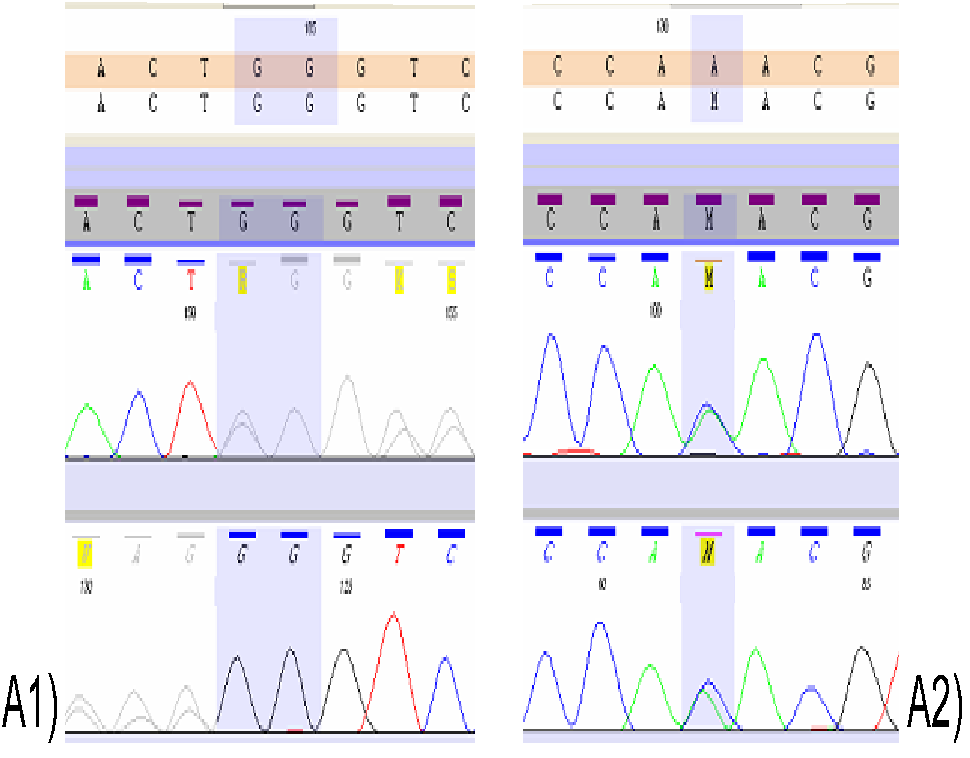
Figure 4: Mutational analysis of ENAM in proband`s parents. A1) p.Leu420_Gly421fs - mutational analysis in the proband`s mother by Sanger method; A2) p.Lys111Thr - mutational analysis in the proband`s father by Sanger method.
Discussion
We describe a case of 6.5 girl in whom the co-occurrence of ENAMel hypoplasia and skeletal open bite lead to preliminary diagnosis of amelogenesis imperfecta. Genetic research of patient revealed the presence of two heterozygous mutations in the ENAM gene (p.Lys111Thr/c.332A>C and p.Leu420_Gly421fs/c.1260_1261insAG). The detected second mutation (p.Leu420_Gly421fs/c.1260_1261insAG) had already been described as a mutation associated with amelogenesis imperfect[14]. Interestingly, p.Leu420_Gly421fs appears to be relatively frequent among the AI patients although this conclusion is apparent only after nomenclature differences used by various authors are taken into account (the same mutation was described as: p.P422fsX448/c.1258–1259 ins AG/g.13185–13186insAG[12,15,16] or p.P422fsX448/c.1259_1260insAG/g.13185_13186insAG[17] or p.V422PfsX448/ c.1259_1230insAG/g.13942_13943/ins AG[3].
Sequencing of the ENAM gene in patient’s parents has shown the father to be a heterozygous carrier of the p.Lys111Thr mutations, and mother a heterozygous carrier of p.Leu420_Gly421fs. However, on the clinical examination no irregularities in the parents’ teeth were found. Our findings add to the previously observed intra and interfamilial phenotypic variation (from normal to profound phenotype) associated with heterozygous ENAM mutations, including p.Leu420_Gly421fs. In AI families described by both Hart et al.[12] and Ozdemir et al.,[15] all heterozygous carriers (N=10) of the p.Leu420_Gly421fs mutation had mild but discernible symptoms (localized hypoplastic ENAMel pitting defects with no open bite). In another study, Pavlic et al.[16] reported a proband with single p.Leu420_Gly421fs mutation associated with extremely mild phenotype of chalky white hypo maturated ENAMel with only mild local hypoplastic alteration, while his father who also carried the mutation had clear local hypoplastic AI phenotype with typical horizontal hypoplastic grooves. Five apparently unaffected individuals carrying heterozygous ENAM p.Leu420_Gly421fs mutation, similar to our proband’s mother, were also described[15].
The severe symptoms of AI seem to occur due to biallelic mutations in the ENAM gene, both homozygous p.Leu420_Gly421fs mutation[15,18] and compound heterozygous for a p.Leu420_Gly421fs mutation in one allele in combination with another mutation in the other allele[15,17]. Our patient was compound heterozygous for mutations in the ENAM gene, while her parents had only one mutant allele of the ENAM gene with no features of AI in their clinical presentation which is consistent with canonical autosomal recessive inheritance.
Our patient presents skeletal open bite. The occurrence of anterior open bite is reported in about 50% of all AI patients[18]. Kida et al.[19] have shown the appearance of open bite in heterozygous patients with regard to mutations in the ENAM gene. However, most researchers suggest that the open bite is one of the more advanced symptoms amelogenesis imperfecta, and occurs rather in patients who are homozygous for a mutation in the ENAM gene[20]. In keeping with this our proband was compound heterozygous for ENAM mutation and the skeletal open bite was observed clinically. In our case the presence of the p.Lys111Thr/c.332A>C mutation apparently enhanced phenotype manifestation of the p.Leu420_Gly421fs mutation.
In conclusion, we describe a novel mutation in the ENAM gene (p.Lys111Thr) found in trans with the p.Leu420_Gly421fs/ (rs587776588) in a patient with amelogenesis imperfecta. Our report emphasizes the relatively high prevalence of the p.Leu420_Gly421fs among the AI patients and adds to the discussion on phenotypic manifestations (or lack thereof) of carriers of mono- and biallelic ENAM defects.
Conflict of interests: None declared
Acknowledgments: The authors are indebted to family that participated in these studies.
Funding: This study was financially supported from the statutory funds by the Medical University of Warsaw (1MA/N/2015).
References
1. Witkop, C.J. Amelogenesis imperfecta, dentinogenesis imperfecta and dentin dysplasia revisited: problems in classification. (1988) J Oral Pathol 17(9-10): 547-553.
2. Alachioti, X.S., Dimopoulou, E., Vlasakidou, A., et al. Amelogenesis imperfecta and anterior open bite: Etiological, classification, clinical and management interrelationships. (2014) J Orthod Sci 3(1): 1-6.
3. Chan, H.C., Estrella, N.M., Milkovich, R.N., et al. Target gene analyses of 39 amelogenesis imperfecta kindreds. (2011) Eur J Oral Sci 119 Suppl 1: 311-323.
4. Toksavul, S., Ulusoy, M., Türkün, M., et al. Amelogenesis imperfecta: the multidisciplinary approach. A case report. (2004) Quintessence Int 35(1): 11-4.
5. Seymen, F., Lee, K.E., Koruyucu, M., et al. ENAM mutations with incomplete penetrance. (2014) J Dent Res 93(10): 988-992.
6. Wang, X., Zhao, Y., Yang, Y., et al. Novel ENAM and LAMB3 mutations in Chinese families with hypoplastic amelogenesis imperfecta. (2015) PLoS One 10(3): e0116514.
7. Hu, J.C., Yamakoshi, Y. ENAMelin and autosomal-dominant amelogenesis imperfecta. (2003) Crit Rev Oral Biol Med 14(6): 387- 398.
8. Gutierrez, S.J., Chaves, M., Torres, D.M., et al. Identification of a novel mutation in the ENAMalin gene in a family with autosomal-dominant amelogenesis imperfecta. (2007) Arch Oral Biol 52(5): 503-506.
9. Hu, J.C., Hu, Y., Lu, Y., et al. ENAMelin is critical for ameloblast integrity and ENAMel ultrastructure formation. (2014) PLoS One 9(3): e89303.
10. Fukae, M., Tanabe, T. Nonamelogenin components of porcine ENAMel in the protein fraction free from the ENAMel crystals. (1987) Calcif Tissue Int 40(5): 286-293.
11. Masuya, H., Shimizu, K., Sezutsu, H., et al. ENAMelin (ENAM) is essential for amelogenesis: ENU-induced mouse mutants as models for different clinical subtypes of human amelogenesis imperfecta (AI). (2005) Hum Mol Genet 14(5): 575-583.
12. Hart, T.C., Hart, P.S., Gorry, M.C., et al. Novel ENAM mutation responsible for autosomal recessive amelogenesis imperfecta and localised ENAMel defects. (2003) J Med Genet 40(12):900-906.
13. Ploski, R., Pollak, A., Müller, S., et al. Does p.Q247X in TRIM63 cause human hypertrophic cardiomyopathy? (2014) Circ Res 114(2): e2-5.
14. Wright, J.T., Torain, M., Long, K., et al. Amelogenesis imperfecta: genotype-phenotype studies in 71 families. (2011) Cells Tissues Organs 194(2-4): 279-283.
15. Ozdemir, D., Hart, P.S., Firatli, E., et al. Phenotype of ENAM mutations is dosage-dependent. (2005) J Dent Res 84(11): 1036-1041.
16. Pavlic, A., Petelin, M., Battelino, T. Phenotype and ENAMel ultrastructure characteristics in patients with ENAM gene mutations g.13185-13186insAG and 8344delG. (2007) Arch Oral Biol 52(3): 209-217.
17. Chan, H.C., Mai, L., Oikonomopoulou, A., et al. Altered ENAMelin phosphorylation site causes amelogenesis imperfecta. (2010) J Dent Res 89(7): 695-699.
18. Crawford, P.J., Aldred, M., Bloch-Zupan, A. Amelogenesis imperfecta. (2007) Orphanet J Rare Dis 2:17.
19. Kida, M., Ariga, T., Shirakawa, T., et al. Autosomal-dominant hypoplastic form of amelogenesis imperfecta caused by an ENAMelin gene mutation at the exon-intron boundary. (2002) J Dent Res 81(11): 738-742.
20. Hart, P.S., Michalec, M.D., Seow, W.K., et al. Identification of the ENAMelin (g.8344delG) mutation in a new kindred and presentation of a standardized ENAM nomenclature. (2003) Arch Oral Biol 48(8): 589-596.










