
The Mammalian Stress Mechanism (MSM) Explains Radial Artery Spasm (RAS)
Affiliation
North County Dental Surgery Center, San Marcos, California, USA
Corresponding Author
Lewis S. Coleman, North County Dental Surgery Center, San Marcos, California, USA, Tel: 661-900-2390; E-mail: lewis_coleman@yahoo.com
Citation
Coleman, L.S. The Mammalian Stress Mechanism (MSM) Explains Radial Artery Spasm (RAS). (2018) J Anesth Surg 5(1): 81- 94.
Copy rights
© 2018 Coleman, L.S. This is an Open access article distributed under the terms of Creative Commons Attribution 4.0 International License.
Keywords
Stress; Hans Selye; Coagulation; Radial artery spasm; RAS; Thrombosis; Angioplasty; Invasive monitoring
Abstract
Objective: Based on stress theory, this paper proposes a fresh explanation of Radial Artery Spasm (RAS), the nature of which remains unclear.(1)
Data Sources: Published research papers accessed via PubMed
Study Selection: Abstract review
Data Extraction: Computerized Internet Search
Data Synthesis: Fresh information relevant to stress theory derived from unrelated research enabled the description of a testable “mammalian stress mechanism” (MSM) that explains the stress theory proposed by Hans Selye.
Conclusions: MSM activity explains the nature of RAS.
Introduction
Radial artery spasm (RAS) occurs after multiple unsuccessfulattempts to cannulate the radial arteryusing small, short catheters for monitoring and blood sampling, whereupon the radial pulsebecomes impalpable and cannulation becomes futile. Persisting ulnar circulation presumably prevents ischemia, and the artery recovers if left unmolested, butsometimes hours after successful cannulationthe pulse wave degenerates and the pulse becomes impalpable as far proximal as the elbow, particularly in the presence of pathology. Diverse treatments relieve RAS including radial nerve blockade, sympathetic ganglion blockade, warming the extremity, aspirating thrombus from the catheter tip, and flushing the artery with local analgesics, vasodilators, and anticoagulants[1,2].
Interventional radiologists have embraced the RAS acronym to explain the “entrapment syndrome” that occurs after the installation of larger and longer angioplasty catheters via the radial artery, causing painful arterial damage in accord with largercathetersand lesserarterial diameter. RAS can also prevent catheter insertion. Angioplasty RAS is routinely relieved by flushing the artery with heparin or cocktails of nitroglycerin and verapamil that lack neuromuscular effects[3].
As its name implies, RAS is attributed to neuromuscular vasospasm. This seems reasonable becausearteries visibly spasm during angioplasty and surgery; sympathetic blockade relieves RAS; andmedical physiology presumes thatopposing forces of cardiac contractility and muscular vasoconstriction governs blood flow[4]. However, muscular spasm offers a weak explanation of RAS, because intense muscle contraction rapidly depletes ATP, causing obligatory muscle exhaustion and relaxation.
Like other intracellular activities, muscle contraction is energized by ATPase enzymes that require Ca+ and ATP[5]. The sarcoplasmic reticulum releases Ca+ into myocyte cytoplasm to initiate contraction. ATPase then energizes the movement of fibrillar actin strands relative to adjacent myosin strands via a “ratcheting mechanism” to contract the muscle[6,7]. A calcium pump mechanism removes Ca+ from the cytoplasm and sequesters it within the sarcoplasmic reticulum to release the ratcheting mechanism, halt ATPase activity, andenable muscular relaxation. ATP depletion accordingly undermines muscle contraction.
The mitochondrial Krebs Cycle efficiently generates ATP in eukaryotic animal cells, but this necessitates oxygen and glucose, so that ATP generation is limited by tissue perfusion and oxygenation. This is readily observed during intense exercise, where cellular oxygen starvation causes skeletal muscle cells to revert to inefficient anaerobic ATP generation, causing muscle fatigue. Exercise conditioning induces angiogenesis (capillary proliferation) in muscle tissues that enhances oxygen delivery, increases ATP generation, and improves exercise tolerance, but only up to a point[8-11].
Unlike skeletal muscle, vascular smooth muscle contracts slowly and enjoys close proximity to oxygenated blood, but its function nevertheless remains dependent on ATP generation. This is illustrated by rigor mortis, where the circulatory failure of death disrupts oxygen transport and delivery, causing cellular anoxia that halts ATP generation by the Krebs Cycle, so that universal muscle flaccidity ensues in the immediate aftermath of death, including vascular smooth muscle. The resilience of the calcium pump prevents rigor mortis in life. Anaerobic metabolism generates enough ATP to sustain the calcium pump for a few hours after death, but as the failing sarcoplasmic reticulum releases its calcium into the cytoplasm, the ratcheting mechanism locks in place for lack of ATP, causing rigor mortis in all types of muscle, including vascular smooth muscle.
Muscular spasm fails to explain why anticoagulants, which lack neuromuscular effects, can successfully prevent and relieve RAS, or why the incidence of RAS is exaggerated by seemingly unrelated diseases including congestive heart failure, hypertension, diabetes, obesity, and cancer.
Ultrasound detects thrombus in RAS; thrombus aspiration restores monitoring catheter function; and flushing the artery with anticoagulants prevents and relieves angioplasty RAS. These observations suggest that thrombus formation causes the RAS phenomenon, but this explanation is frustrated for lack of an effective hemostasis explanation. Such, however, may no longer be the case, because the recently discovered mammalian stress mechanism clarifies the nature of coagulation and its relationships with nervous activity, tissue repair, and disease[12-17].
Methods
The author’s curiosity about the recently discovered chimeric nature of coagulation factor VIII inspired a six-year review of published research via the Internet using advanced computer techniques. Factor VIII consists of enzymatically inert von Willebrand factor (VWF) and enzymatic factor VIIIC. The vascular endothelium manufactures VWF and releases it into blood circulation under nervous control, while VIIIC is continuously released by the liver. The two gigantic molecules bind together in blood circulation and exert their effects in concert. VWF stabilizes VIIIC and enables its enzymatic effects, which are otherwise so labile as to be nonexistent. Thus the factor VIII chimera links nervous activity to blood enzyme activity. Defects in VIIIC cause true hemophilia, a severe, sex-linked clotting diathesis. Defective VWF causes the von Willebrand coagulation diathesis, which is usually mild but in severe forms can mimic true hemophilia. VWF defects also cause angiodysplasia, because the VWF molecule maintains capillary structural integrity[18-24]. Theunique characteristics of factor VIII served as a “Rosetta Stone” that deciphered the relationships between and among coagulation enzymes, nervous activity, coagulation, capillary hemostasis, atherosclerosis, bleeding diatheses, sickle cell anemia, angiodysplasia, angioneurotic edema, hemodynamic physiology, tissue repair, disease, and stress[25-30].
Results
The literature review successivelyidentifiedtestable mechanisms of coagulation[12,14], capillary hemostasis[15], atherosclerosis[31,33], tissue repair[34], inflammation, apoptosis[15], the surgical stress syndrome[35], anesthesia, analgesia, allostasis[35,36] and a capillary gate mechanism that explains hemodynamic physiology[13]. These seemingly disparate mechanisms were ultimately comprehended as elements of the long-sought “mammalian stress mechanism” (MSM) postulated by Hans Selye[16,37-43]. The MSM is testable, and it enables Selye’s revolutionary “unified theory of medicine”[42] that explains physiology, pathology, stress, and their relationships.
In retrospect, Selye was ahead of his time, like Jules Verne predicting trips to the moon before rockets were invented. He was an endocrinologist, and HPA hormone elevations and gross organ effects were the only recognized reactions to stress in his time[43,44]. The intense international 30 year search for his putative mechanism that followed the discovery of DNA focused on hormones and failed to find the coagulation information needed to identify the elusive mechanism. However, the stress researchers were closer to success than they realized. They developed capillary gate theory and tissue repair theory to facilitate their search, and these hypotheses are embodied in the MSM. Another 30 years of fresh information from unrelated research was needed. Selye would have been surprised to learn that coagulation enzymes are the focus of his theory, that von Willebrand factor (VWF) is the prototypical stress hormone, and that his mechanism confers a unified theory of biology that exceeds the bounds of medicine. These implications will be elaborated in an upcoming book that is in the hands of its publisher.
This presentation will briefly review the MSM and its operation, which explains the nature of RAS.
The Mammalian Stress Mechanism (MSM)
The mammalian stress mechanism (figure 2) is analogous to the familiar coagulation cascade (figure 1), but it incorporates recent research that explains its relationships to nervous activity and tissue repair.It appears during the early stages of embryological development, and it converts chromosomal genetic information into cell specialization and cell organization that creates complex multicellular anatomical structures. It remains continuously active for the duration of life to regulate hemodynamic physiology, maintain the “internal milieu” that optimizes cell survival and function[45], and maintain mature structures. Meanwhile, DNA resumes quiescence once embryological development is complete. Stress-induced MSM hyperactivity causes disease manifestations, and explains the relationships of diseases and stresses. For example, it explains why diabetes, hypertension, obesity, malignancy, and congestive heart failure are closely associated, and why seemingly disparate diseases commonly manifest fever, inflammation, edema, cachexia, and malaise. It explains the relationships of hemodynamic physiology, coagulation, atherosclerosis, tissue maintenance, and tissue repair, and thereby confers a fresh, simplified explanation of the puzzling RAS phenomenon[16,17,34].
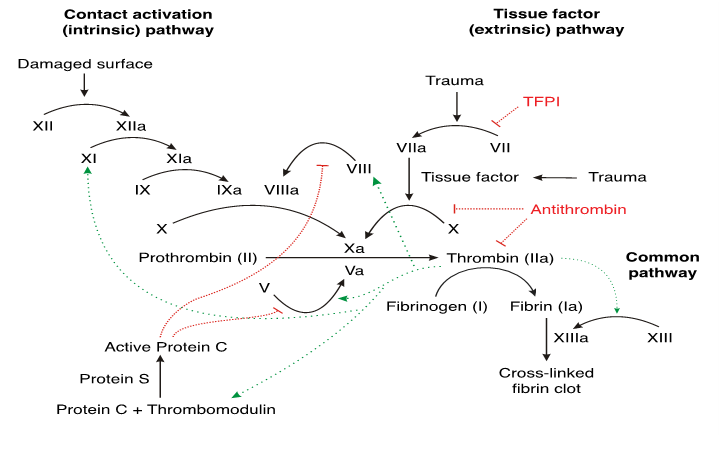
Figure 1: The Coagulation Cascade was described in the early 1960’s. It explains coagulation as a “cascade” of enzymatic interactions that produces insoluble fibrin, but it fails to explain how the cascade is initiated, regulated, and concluded. It assumes that the sole purpose of coagulation is hemostasis. Conventional “vasoconstriction” theory fails to explain capillary hemostasis, because capillaries lack the ability to contract. The cascade consists of an “intrinsic pathway” consisting of factors VIII and IX, and an “extrinsic pathway” consisting of factor VII and tissue factor. Both pathways interact with factor X in a “final common pathway” to generate thrombin, soluble fibrin, and insoluble fibrin. By Joe D - Own work, CC BY-SA 3.0,https://commons.wikimedia.org/w/index.php?curid=1983833
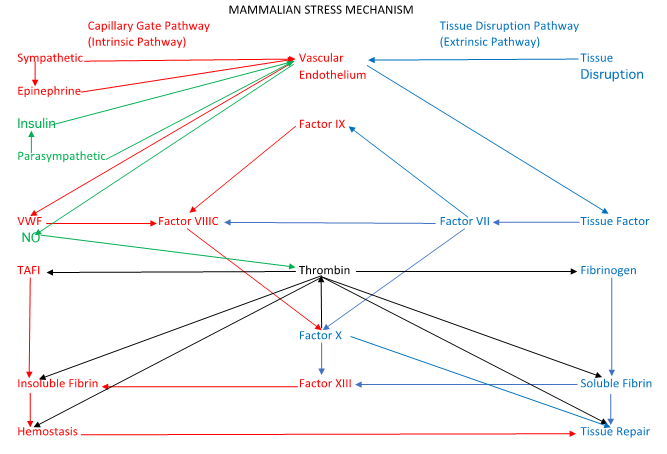
Figure 2: A simplified diagram of the “Mammalian Stress Mechanism” (MSM).
The MSM is analogous to the coagulation cascade, but it incorporates recent research that clarifies the relationships of stress, nervous activity, tissue disruption, tissue repair, hemostasis, and hemodynamic physiology. Sympathetic tone releases VWF from the vascular endothelium to activate the “capillary gate component” of the MSM, shown in red, which is analogous to the “intrinsic pathway” of the coagulation cascade. Parasympathetic tone, shown in green releases nitric oxide (NO) from the vascular endothelium to open the capillary gate. Tissue damage activates the “tissue disruption pathway,” shown in blue that is analogous to the “extrinsic pathway” of the coagulation cascade, to enable tissue repair.
The Three Products of the MSM
Combinations of nervous activity and tissue disruptionalter the enzymatic interaction of factors VII, VIII, IX and X to regulate the magnitude, location, and speed of production of the three MSM products, which are thrombin, soluble fibrin, and insoluble fibrin. This focuses MSM activity to regulate hemodynamic physiology, hemostasis, and tissue repair. Stress induces MSM hyperactivity thatproduces these three products in excess, which manifests as disease. The constantly fluctuating levels of the three products producesa bewildering blizzard of symptoms and manifestations that belie the relative simplicity of MSM operation.
Thrombin is the “universal enzyme of extracellular energy transduction.” Like intracellular ATPase enzymes, it requires Ca+ and ATP, and it transforms ATP energy into action[46-58].
Thrombin is generated when tissue factor, or TF (a glycoprotein in extravascular tissues), and Factor VII (a blood enzyme) meet in the presence of prothrombin. The selectively permeable vascular endothelium allows small quantities of TF to “leak” into blood circulation, and it allows small quantities of factor VII to “penetrate” into extravascular tissues. This generates small quantities of thrombin throughout the body (from its precursor, prothrombin). This “background” thrombin generation energizesongoing tissue maintenance and a capillary gate mechanism regulated by autonomic balance that governs hemodynamic physiology[59,60].
When trauma disrupts the vascular endothelium (even minor damage at the capillary level), the MSM accelerates thrombin generation in those damaged tissues to energize hemostasis[61]. The MSM then maintains thrombin elevations within an optimal range to energize cellular tissue repair activities. It reduces thrombin generation to maintenance levels in healing tissues as the repair process nears completion, causing clot disintegration and apoptosis that shrinks granulation tissues to enable wound closure[62,63].
Parathyroid glands regulate extracellular Ca+ within a narrow range that optimizes thrombin activity[24,64-77]. Drugs and chemicals that elevate Ca+ levels exaggerate thrombin generation, and vice-versa. Mg+ competitively inhibits Ca+ and mitigates thrombin generation[27,64,78-94]. Magnesium sulfate is used to treat eclampsia, a treatment readily explained by its thrombin inhibition.
Thrombin is essential for embryological development, tissue maintenance, tissue repair and for malignancy. Pharmaceutical effects and enzyme defects that inhibit thrombin generation also disrupt embryological development, tissue maintenance, tissue repair, and malignancy.
All cells thus far tested have PAR (thrombin receptors) on their outer surface that determine how they react to thrombin elevations. Four different types of PAR have been discovered. Individual cell types have characteristic PAR types and numbers that determine how the cell reacts to thrombin elevations. Like sails on tiny ships, these can be reconfigured by the cell to alter cell reactions to thrombin during embryological development, tissue repair, and malignancy[57,62,63,95-102].
Ordinarily thrombin elevations energize cellular activities and inhibit apoptosis while thrombin starvation initiates apoptosis in fibroblasts, but cells react to thrombin differently at different times and in different contexts. Thrombin can energize apoptosis, inhibit apoptosis, or be toxic to the cell. Additional research is needed to clarify thrombin effects, as present understanding relies on invitro studies that may not apply to living organism.
The following thrombin effects of thrombin reflect its universal role in extracellular ATP energy utilization.
• Chemotaxis of platelets, osteocytes, white blood cells, and other tissue repair cells[69,103-105]
• Mitosis[59,69,102,106]
• Metabolism[69]
• Hypertrophy[69,105,107-110]
• Angiogenesis[47,111,112]
• Platelet activation, chemotaxis, and thromboxane release[46]
• Proliferation, spreading and gap formation in the vascular endothelium[117,118]
• Chemokine, cytokine, interleukin, bradykinins, caspase, and prostaglandin release[96,104,109,119-127]
• Bone, muscle, collagen, and immune protein production by osteocytes, myocytes, fibroblasts, and immune cells [54,63,66,105,106,108,110,118,128-131,132,133-139]
• Conversion of fibrinogen to soluble fibrin[72]
• Conversion of fibrillar soluble fibrin to three-dimensional insoluble fibrin[61,71,140-148]
• Stabilization of insoluble fibrin via “Thrombin-Activated Fibrinolysis Inhibitor” (TAFI) [145,149-152]
• Inflammation, which dissolves the “basement membrane” that binds cells in tight formation with one another and with the Vascular Endothelium to facilitate chemotaxis[66,98].
• Astrocyte and glial cell proliferation in brain tissue[102].
• Gelsolin activation[153,154]
• Complement activity[155] Complement cascade activity generates large amounts of thrombin.
• T-cell activation[99,133]
• Blast transformation in lymphocytes[131,133]
• Macrophage phagocytic activity[67,99,111,112,131,136,156]
• Plasma (immune) cell and neutrophil activation[136,147,157]
• “Tumor Necrosis Factor” release from microglial cells[158]
• Tumor growth, malignancy, and fibrosis[55,62,63,95,128,130,135,159,160,161]
• Inhibits apoptosis[57,95,100,101,162,163]
• Intracellular gap formation in the vascular endothelium[117]
• Defects in Factors VII, X and tissue factor lethally disrupt thrombin generation[164]
Soluble fibrin is the “universal protein of tissue repair.” Thrombin converts fibrinogen to soluble fibrin that appears in pus, exudates, scabs, scars, saliva, mucus, and milk[165,166]. It escapes the vascular system via inflammatory gaps in the vascular endothelium andinfiltrates damaged tissues to promote fibroblast proliferation and collagen production that facilitates tissue repair[106,117,165]. Excessive insoluble fibrin causes tissue edema, organ dysfunction, fibrosis, and scar formation[139,167-178].
Insoluble fibrin is the “universal polymer of hemostasis.” Factor VIII accelerates thrombin generation to energize its enzymatic conversion of soluble fibrin into strands of insoluble fibrin that entangle blood cells, reduce pulsatile turbulence below a threshold, and bind blood cells into a viscoelastic clot or thrombus[12,33,56]. In capillaries, insoluble fibrin exaggerates flow resistance to regulate hemodynamic physiology[13]. Excessive insoluble fibrin exaggerates blood viscosity and coagulability, which decreases cardiac output, tissue perfusion, and tissue oxygenation, and invites infarction, thrombosis, embolism, and disseminated intravascular coagulation (DIC)[35]. Excessive insoluble fibrin generation exhausts clotting precursors[179,180].
Insoluble fibrin incorporates cross-links of plasminogen, which spontaneously degrades into plasmin that enzymatically disintegrates insoluble fibrin into “fibrin split products.” Thrombin-activated fibrinolysis inhibitor (TAFI) stabilizes plasminogen and preserves insoluble fibrin[145,149-152].
The Interaction of factors VII, VIII, IX, and X
Hepatic enzyme factors IX and X have prolonged half-lives and circulate at stable levels, but factors VII and VIIIC are labile, so that their fluctuating enzymatic activities alter the enzymatic interaction and determine the rate, magnitude, location and speed of production of thrombin, soluble fibrin, and insoluble fibrin.
Factor IX enhances factor VIII activity but lacks other effects
Factor VIII links nervous activity to blood enzymes. It is a gigantic chimeric molecular complex consisting of continuously released hepatic enzyme factor VIIIC and von Willebrand factor (VWF) that is produced by the vascular endothelium and released into blood in accord with sympathetic activity[29]. These seemingly unrelated molecules bind together in blood circulation and exert their effects in concert, so that factor VIII fluctuates in accord with nervous activity, including emotion[26,29,181-183]. Factor VIII interacts with factors IX and X to generate factor XIII that adds “cross links” of fibronectin, vitronectin and plasminogen to molecular strands of soluble fibrin to generate insoluble fibrin in capillaries and flowing blood.
Factor VII links tissue damage to blood enzymes. Tissue damage disrupts the vascular endothelium and exposes factor VII to tissue factor in extravascular tissues[184]. Tissue factor stabilizes labile factor VII, whereupon it generates small amounts of thrombin that enable the activities of factors VIII, IX, and X. Factor VII thus functions as a “trigger” that initiates and localizes the enzymatic interaction.
The pivotal activities of factor X have yet to be fully elucidated. It interacts with factor VII and tissue factor to enable embryological development and tissue repair, and it interacts with factor VIIIC and VWF (factor VIII) to generate insoluble fibrin that enables hemostasis and capillary gate function.
The Vascular Endothelium
The vascular endothelium is a diaphanous layer of cells, one cell thick, that lines the inner surface of blood vessels and is the sole substance of capillaries. It regulates the enzymatic interaction of factors VII, VIII, IX and X to govern the rate, magnitude, and location of the production of thrombin, soluble fibrin, and insoluble fibrin.
The vascular endothelium insulates blood enzymes from tissue factor in extravascular tissues. Its traumatic disruption exposes tissue factor to blood enzymes and initiates coagulation and tissue repair. Harmful radiation and toxic chemicals increase its permeability to factors VII, X and tissue factor, which causes painful inflammation but does not induce coagulation because the intact vascular endothelium remains impermeable to gigantic factor VIII. Abundant tissue factor exaggerates coagulability and malignancy in brain, nerves, lung, gonads, arteries, cervix, and placenta[184].
The vascular endothelium is “selectively permeable.” It allows the continuous “penetration” of factor VII from flowing blood into extravascular tissues, which generates small amounts of thrombin that energize tissue maintenance. It allows the continuous “leakage” of tissue factor into flowing blood, which enables the continuous “background” activity of factors VII, VIII, IX and X.
The cells of the vascular endothelium react to local factors and communicate with one another via electromagnetic signals[185]. They release VWF in accord with sympathetic tone to generate insoluble fibrin[26,186-190]. They release nitric oxide (NO) in accord with parasympathetic tone to disintegrate insoluble fibrin (nitrergic neurogenic vasodilation)[189,191-195].
The Tissue Disruption Pathway
The tissue disruption pathway is analogous to the extrinsic pathway of the coagulation cascade. Tissue damage disrupts the ubiquitous vascular endothelium, exposes tissue factor to blood enzymes, and triggers an intense enzymatic interaction of factors VII, VIII, IX and X that activates platelets, releases thromboxane, and generates strands of insoluble fibrin that entangle blood cells, reduce pulsatile blood turbulence below a threshold, and bind blood cells into a viscoelastic clot that restores the isolation of damaged tissues from flowing blood[12].
The Tissue Repair Mechanism
Due to its gigantic size, factor VIII cannot penetrate the clot of its own manufacture, and factor IX interacts only with factor VIII, so that clot formation is limited to the vicinity of tissue damage. The selectively permeable viscoelastic clots regulates the penetration of factors VII and X into damaged tissues, where they interact with tissue factor to generate thrombin that energizes inflammatory gaps between the cells of the vascular endothelium that increase its permeability. Thrombin energized inflammation loosens cell connections to facilitate thrombin energized chemotaxis of repair cells that move from adjacent undamaged into damaged tissues, where they engage in thrombin energized tissue repair. Thrombin-generated soluble fibrin escapes the vascular system through thrombin inflamed tissues to enter damaged tissues, where it facilitates thrombin energized fibroblast proliferation and collagen production that fills empty spaces. Thrombin energized immune activity fights infection and removes debris. Thrombin energized cell differentiation replaces damaged bone and tissues. Thrombin generation declines as tissue repair restores the vascular endothelium, and thrombin starvation induces clot disintegration and repair cell apoptosis that draws wound edges together to conclude the repair process[95].
The Capillary Gate Pathway
The capillary gate pathway is analogous to the intrinsic pathway of the coagulation cascade. Nervous activity releases von Willebrand factor (VWF) from the vascular endothelium into flowing blood to stabilize VIIIC and generate insoluble fibrin that increases blood viscosity and coagulability.
The Capillary Gate Mechanism
Capillary surface area is vastly greater than that of all larger vessels combined, and turbulence, flow rates and pressures are minimal at the capillary level. The capillary gate pathway regulates a submicroscopic “capillary gate mechanism” that governs capillary flow, capillary hemostasis, systemic vascular resistance, tissue perfusion, organ function, cardiac output, cardiac efficiency, blood pressure, and pulse rate. Autonomic balance and CO2 tissue accumulation regulate the capillary gate mechanism[196].
Sympathetic activity extrudes VWF from the inner walls of capillaries, next to binding sites for fibrinogen and fibronectin. Factor VIIIC binds to VWF and accelerates thrombin generation to convert fibrinogen to strands of soluble fibrin. Factor VIII then converts factor X to factor XIII that adds “cross-links” of plasminogen and fibronectin to molecular strands to soluble fibrin to generate insoluble fibrin that polymerizes into strands that “close” the capillary gate by increasing capillary flow resistance[73,180]. Plasminogen spontaneously degenerates into plasmin that enzymatically disintegratesinsoluble fibrin into “fibrin split products” unless plasminogen is continuously stabilized by “thrombin-activated plasminogen inhibitor” (TAFI). Parasympathetic activity releases nitric oxide (NO) from the vascular endothelium[189,191-195]. NO is a gaseous molecule that diffuses into the capillary lumen and binds to Ca+, which inactivates thrombin, accelerates the disintegration of insoluble fibrin, and “opens” the capillary gate (aka “nitrergic neurogenic vasodilation”).
The opposing effects of epinephrine and insulin extend autonomic balance to peripheral tissues where direct autonomic innervation is lacking. Sympathetic activity releases epinephrine from the adrenal glands, which releases VWF from the vascular endothelium, increases factor VIII activity, and generates insoluble fibrin[197]. Parasympathetic activity releases insulin from the pancreas, which releases NO from the vascular endothelium, which accelerates insoluble fibrin disintegration[198].
The Turbulence Mechanism
Familiar fluids such as water, oil, steam, and atmospheric gases are classified as “Newtonian” because they exhibit exponential increases in turbulent flow resistance when they are accelerated in pipes[199]. (see figure 3) In contrast, blood is a “non-Newtonian” fluid that exhibits exponential decreases in flow resistance when it is accelerated in arteries. This is because mammalian red cells spontaneously form “aggregates” during blood acceleration that inhibit turbulent flow resistance, which enables the heart to efficiently eject its contents in less than a tenth of a second[200]. The muscular arterial tree expands to accommodate cardiac ejection volume, and then functions as a “secondary heart” that propels blood toward capillary beds as it restores resting volume. However, blood flow momentarily reverses direction in the aorta at the outset of diastole, which closes the aortic valve. The reduced diameter of the distal aorta amplifies the momentary flow reversal, disrupts the aggregate patterns, and produces a burst of diastolic pulsatile turbulence that momentarily halts blood flow as it propagates toward the periphery of the arterial tree. Laminar blood flow resumes in the wake of the pulse wave. The pulsatile turbulence generates lateral forces that press on the inner walls of arteries. This explains blood pressure and the palpable pulse. The turbulence maintains arterial patency by disintegrating thromboses and mobilizing particulate deposits from the inner walls of arteries that would otherwise induce localized inflammation and tissue repair activities that cause atheroma formation. Decreasing arterial diameter exaggerates turbulence, which explains why atherosclerosis and thrombosis are rare in distal extremities[12,31,32].
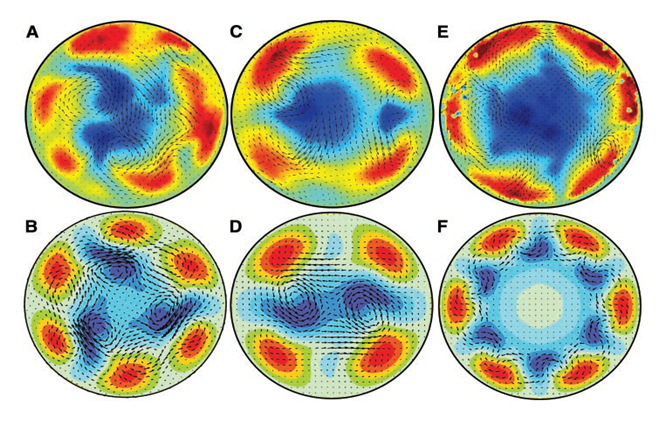
Figure 3: Newtonian Pipe Flow Turbulence.(199) Turbulent forward flow appears as fast-moving “jet streams” (shown in red) that form along the inner walls of pipes and force slow-moving fluid to the center, where it moves backwards (shown in blue), causing increased viscosity (flow resistance). A, C and E are laser photographs that reflect flow acceleration that exaggerates turbulent intensity. B, D and F are computer simulations. Similar diastolic turbulence inhibits atherosclerosis by mobilizing particulate deposits from the inner walls of arteries. Diastolic turbulence also generates lateral forces that explain blood pressure and the palpable pulse. The study of Hof et al has revolutionized the understanding of pipe flow turbulence and viscosity and refuted several older concepts that attempted to explain the manifestations of fluid flow in pipes and arteries. This includes systemic blood pressure (BP), pulmonary artery pressure (PAP), Pulmonary Vascular Resistance (PVR), the Hagen-Poiseuille Equation, Mean Arterial Pressure (MAP), Shear Stress, and Reynolds Numbers. (Reproduced with permission Science).
Accelerated Capillary Senescence
Capillary gate activity regulates tissue perfusion, but does not normally cause tissue oxygen starvation, as evidenced the by the rarity of infarction in youth. However, inexorable capillary deterioration proceeds with senescence, which inexorably undermines tissue perfusion and glucose uptake, exaggerates flow resistance that causes essential hypertension, harmfully increases cardiac work that induces congestive heart failure, and alters pulsatile turbulence in favor of lateral forces at the expense of turbulent intensity that maintains arterial patency. Chronic stress, including toxic chemicals, smoking, fear, anxiety, obesity, and chronic illness, accelerates capillary senescence and promotes hypertension, diabetes, congestive heart failure, infarction, eclampsia, and multi-organ system failure. I call this “accelerated capillary senescence[201-205].” Exercise conditioning mitigates capillary senescence by inducing angiogenesis[8-11,206,207].
Discussion
The MSM clarifies RAS, as follows:
Malignancy, obesity, smoking, emotional stress[81,83,208], hypertension, and other illnesses induce MSM hyperactivity that exaggerates blood viscosity and coagulability, accelerates capillary senescence, and promotes thrombus formation. Capillary senescence exaggerates flow resistance, which undermines turbulent intensity, causes essential hypertension, promotes thrombus formation, accelerates atherosclerosis, c and exaggerates cardiac work, which and causes congestive heart failure that further undermines pulsatile turbulent intensity. In reasonably healthy individuals, pulsatile blood turbulence maintains arterial patency in the aftermath of arterial cannula installation. However, co-existing disease inhibits turbulent intensity, which promotes thrombus formation and propagation that undermines the pulse wave and obstructs arterial flow hours later. This explains the close relationships of RAS and disease.
Nerve blockade promotes spontaneous thrombus disintegration by inhibiting sympathetic nervous activity that releases VWF from the vascular endothelium.
The unappreciated anticoagulant properties of local analgesics such as lidocaine[209-211], calcium channel blockers such as Verapamil, beta-blockers such as propranolol[212], furosemide[213], and other pharmaceuticals explains their ability to relieve RAS.
Multiple arterial piercings increase tissue factor exposure and trigger thrombus formation that mimics spasm and obstructs arterial flow. Soon thereafter, pulsatile turbulence “tunnels” through the thrombus, restores arterial patency, and mimics spasm relief.
Blood is ordinarily transparent to both X-rays and ultrasound, but pulsatile turbulence reflects the Doppler ultrasound signal, which facilitates cannula installation. Doppler ultrasound detects mature thrombus formation but cannot detect immature thrombus formation that nevertheless undermines the palpable pulse and blood flow.
Angioplasty catheters disrupt the vascular endothelium along their entire length and cause far greater tissue factor exposure than small monitoring catheters. This induces thrombus formation along the length of the catheter in accord with small arterial diameter, large catheter diameter, and catheter length. The viscoelastic thrombus is tough, sticky, and flexible, and it bonds to the catheter, causing entrapment. The entrapment eventually resolves as plasmin degrades the viscoelastic clot, but ensuing tissue repair activity causes permanent arterial damage. Anticoagulant heparin, lidocaine, and verapamil infusions prevent and relieve catheter entrapment via hemolysis.
Cold exaggerates blood viscosity and coagulability[214]. Warming the extremity reduces blood viscosity and coagulability, accelerates thrombus disintegration, and restores the waveform.
Ultrasound releases NO from the vascular endothelium, disrupts insoluble fibrin, disintegrates the thrombus, and restores the waveform[215-217].
The MSM suggests simple, safe, inexpensive RAS treatments that can be synergistically combined. Ultrasound releases NO from the vascular endothelium and disintegrates insoluble fibrin. CO2 supplementation of inhaled gas mixtures opens the capillary gate and optimizes pulsatile turbulence intensity. EDTA, trisodium citrate, and MgSO4 are more potent than heparin, and they can be readily reversed with Ca+. They are inexpensive, and they have excellent safety records when used for chelation, dialysis, eclampsia, and blood preservation.
Conclusion
MSM activity here in does not refute spasm, but it offers an alternative explanation of RAS that invites further investigation. It is possible that a combination of spasm and coagulation causes the confusing manifestations of RAS. On the other hand, the Razor of Occam suggests that the simplest explanation is the one most likely to be correct. The coagulation hypothesis is simpler because it potentially explains all aspects of the RAS phenomenon.
The implications of the MSM exceed the bounds of medicine. In addition to enabling Selye’s “unified theory of medicine” that explains physiology, pathology and stress, it confers a “unified theory of biology” that explains embryology, evolution, anatomy, ethology, intelligence, emotion, taxonomy, paleontology, dinosaurs, the Cambrian explosion, and the origin of life. It paves the path for understanding of the gene code, with implications that presently remain in the realm of science fiction. A book that discusses the extended medical and biological implications of stress theory is in the hands of its publisher and will soon be announced via my website: www.stressmechanism.com.
Conflict of Interest: No financial support was provided to produce this paper.
Acknowledgment: The viewpoint presented in this paper is exclusively that of the author.
References
- 1. Bhakta, P., Zaheer, H. Ultrasound-guided radial nerve block to relieve cannulation-induced radial arterial spasm. (2017) Can J Anaesth 64(12): 1269-1270.
- 2. Backman, S.B. Radial artery spasm: Should we worry? (2017) Can J Anaesth 64(12): 1165-1168.
- 3. Kristic, I., Lukenda, J. Radial artery spasm during transradial coronary procedures. (2011) J Invasive Cardiol 23(12): 527-531.
PubMed||CrossRef||Others
- 4. Cannon, W.B. The wisdom of the body. (1932) (W.W. Norton & Company, New York,, 1939) xviii, 19-333.
PubMed||CrossRef||Others
- 5. Ferjani, I., Fattoum, A., Manai, M., et al. Two distinct regions of calponin share common binding sites on actin resulting in different modes of calponin-actin interaction. (2010) Biochim Biophys Acta 1804(9): 1760-1767.
- 6. Hanson, J. Huxley, H.E. Structural basis of the cross-striations in muscle. (1953) Nature 172(4377): 530-532
- 7 Huxley, H. Hanson, J. Changes in the cross-striations of muscle during contraction and stretch and their structural interpretation. (1954) Nature 173(4412): 973-976.
- 8. Ades, P.A., Waldmann, M.L., Meyer, W.L., et al. Skeletal muscle and cardiovascular adaptations to exercise conditioning in older coronary patients. (1996) Circulation 94(3): 323-330.
- 9. Prior, B.M., Lloyd, P.G., Yang, H.T., et al. Exercise-induced vascular remodeling. (2003) Exerc Sport Sci Rev 31(1): 26-33.
- 10. Wijnen, J.A., Kool, M.J.F., van Baak, M.A., et al. Effect of exercise training on ambulatory blood pressure. (1994) Int J Sports Med 15(1): 10-15.
- 11. Zanesco, A., Antunes, E. Effects of exercise training on the cardiovascular system: Pharmacological approaches. (2007) Pharmacol Ther 114(3): 307-317.
- 12. Coleman, L.S. Insoluble fibrin may reduce turbulence and bind blood components into clots. (2005) Med Hypotheses 65(4): 820-821.
- 13. Coleman, L.S. A capillary hemostasis mechanism regulated by sympathetic tone and activity via factor VIII or von Willebrand’s factor may function as a “capillary gate” and may explain angiodysplasia, angioneurotic edema, and variations in systemic vascular resistance. (2005) Med Hypotheses 66(4): 773-775.
- 14. Coleman, L.S. To the Editor: Is von Willebrand Factor a Hormone that Regulates a Coagulation Mechanism? (2006) World J Surg 30(3): 479-481.
- 15. Coleman, L.S. A capillary hemostasis mechanism regulated by sympathetic tone and activity via factor VIII or von Willebrand’s factor may function as a “capillary gate” and may explain angiodysplasia, angioneurotic edema, and variations in systemic vascular resistance. (2006) Med Hypotheses 66: 773-775.
PubMed||CrossRef||Others
- 16. Coleman, L.S. A Stress Repair Mechanism that Maintains Vertebrate Structure during Stress. (2010) Cardiovasc Hematol Disord Drug Targets 10(2): 111-137.
- 17. Coleman, L.S. in Hypotheses in Clinical Medicine, e. a. Shoja MM, Ed. (Nova Biomedical, New York, NY, 2012) chap. 29.
PubMed||CrossRef||Others
- 18. Drenth, J.P., Nagengast, F.M. Aortic stenosis and intestinal blood loss from angiodysplasia: valve replacement is a therapeutic option. (2000) Ned Tijdschr Geneeskd 144: 2237-2240.
PubMed||CrossRef||Others
- 19. Fujita, H., Tomiyama, J., Chuganji, Y., et al. Diffuse angiodysplasia of the upper gastrointestinal tract in a patient with hypertrophic obstructive cardiomyopathy. (2000) Intern Med 39(5): 385-388.
- 20. O’Brien, J.R. Angiodysplasia, haemostasis and capillaries. A hypothesis. (1996) Thromb Res 84(5): 385-387.
- 21. Rosborough, T.K., Swaim, W.R. Acquired von Willebrand’s disease, platelet-release defect and angiodysplasia. (1978) Am J Med 65(1): 96-100
- 22. Tomori, K., Nakamoto, H., Kotaki, S., et al. Gastric angiodysplasia in patients undergoing maintenance dialysis. (2003) Adv Perit Dial 19: 136-142.
PubMed||CrossRef||Others
- 23. Veyradier, A., Balian, A, Wolf, M., et al. Abnormal von Willebrand factor in bleeding angiodysplasias of the digestive tract. (2001) Gastroenterology 120(2): 346-353.
- 24. Warkentin, T.E., Moore, J.C., Anand, S.S., et al. Gastrointestinal bleeding, angiodysplasia, cardiovascular disease, and acquired von Willebrand syndrome. (2003) Transfus Med Rev 17(4): 272-286.
- 25. Bloom, A.L. The biosynthesis of factor VIII. (1979) Clin Haematol 8(1): 53-77.
PubMed||CrossRef||Others
- 26. Federici, A.B. The factor VIII/von Willebrand factor complex: basic and clinical issues. (2003) Haematologica 88(6): EREP02.
PubMed||CrossRef||Others
- 27. Sakariassen, K.S., Ottenhof-Rovers, M., Sixma, J.J. Factor VIII-von Willebrand factor requires calcium for facilitation of platelet adherence. (1984) Blood 63(5): 996-1003.
- 28. Mannucci, P.M., Lobina, G.F., Ruggeri, Z.M. Alterations in fibrinolysis and blood coagulation. (1969) Lancet 293(7592): 466.
- 29. Mannucci, P.M., Ruggeri, Z.M., Gagnatelli, G. Nervous regulation of factor-VIII levels in man. (1971) Br J Haematol 20(2): 195-207.
- 30. Sadler, J.E., Budde, U., Eikenboom, J.C., et al. Update on the pathophysiology and classification of von Willebrand disease: a report of the Subcommittee on von Willebrand Factor. (2006) J Thromb Haemost 4(10): 2103-2114.
- 31. Coleman, L.S. An Improved Explanation of Atherosclerosis. (2005) PLoS Medicine 2(4): e98.
PubMed||CrossRef||Others
- 32.Coleman, L.S. Atherosclerosis may be caused by inadequate levels of turbulence and mixing. (2006) World J Surg 30: 638-639.
PubMed||CrossRef||Others
- 33. Coleman, L.S. A Hypothesis: Factor VII governs clot formation, tissue repair and apoptosis. (2007) Med Hypotheses 69(4): 903-907 .
- 34. Coleman, L.S. Stress repair mechanism activity explains inflammation and apoptosis. (2012) Advances in Bioscience and Biotechnology 3: 459-503.
PubMed||CrossRef||Others
- 35. Coleman, L.S. A testable hypothesis that may explain the morbidity and mortality caused by surgical stress. (2006) Anesth Analg 103(6): 1589; author reply 1589-1590.
- 36. Coleman, L.S. 30 Years Lost in Anesthesia Theory. (2012) Cardiovasc Hematol Agents Med Chem 10(1): 31-49.
- 37. Coleman, L.S. A Testable Stress Mechanism Explains Both Unified Stress Theory and Capillary Gate Theory. FDA Anesthesia & Life Support Drugs Advisory Committee pp239-243 3/29/07, (2007).
PubMed||CrossRef||Others
- 38. Selye, H. The general adaptation syndrome and the diseases of adaptation. (1946) J Clin Endocrinol Metab 6: 117-230.
- 39. Selye, H. Stress and the general adaptation syndrome. (1950) Br Med J 1(4667): 1383-1392.
- 40. Selye, H. Annual Report on Stress. The Physiology and Pathology of Exposure to Stress: A Treatise Based on the Concepts of the General-adaptation-syndrome and the Diseases of Adaptation. (1951) Quarterly Rev of Biol 26(4).
PubMed||CrossRef||Others
- 41. Selye, H. The general adaptation syndrome as a basis for a unified theory of medicine. (1952) Oral Surg Oral Med Oral Pathol 5(4): 408-413.
PubMed||CrossRef||Others
- 442. Selye, H. Sketch for a unified theory of medicine. (1954) Int Rec Med Gen Pract Clin167: 181-203.
PubMed||CrossRef||Others
- 43. Selye, H. A syndrome produced by diverse nocuous agents. 1936. (1998) J Neuropsychiatry Clin Neurosci 10(2): 230-231.
- 44. Selye, H. Thymus and adrenals in the response of the organism to injuries and intoxications. (1936) Br J Exp Pathol 17(3): 234-224.
- 45. Bernard, C. An introduction to the study of experimental medicine. (1949) Schuman, New York: 226.
PubMed||CrossRef||Others
- 46. Sambrano, G.R., Weiss, E.J., Zheng, Y.W., et al. Role of thrombin signalling in platelets in haemostasis and thrombosis. (2001) Nature 413(6851): 74-78.
- 47. Archiniegas, E., Neves, C.Y., Candelle, D., et al. Thrombin and its protease-activated receptor-1 (PAR1) participate in the endothelial-mesenchymal transdifferentiation process. (2004) DNA Cell Biol 23(12): 815-825.
- 48. Armstrong, M.T., Lee, D.Y., Armstrong, P.B. Regulation of proliferation of the fetal myocardium. (2000) Dev Dyn 219: 226-236.
- 49. Murakami, K., Ueno, A., Yamanouchi, K., et al. Thrombin induces GRO alpha/MGSA production in human umbilical vein endothelial cells. (1995) Thromb Res 79(4): 387-394.
- 50. Moser, M., Patterson, C. Thrombin and vascular development: a sticky subject. (2003) Arterioscler Thromb Vasc Biol 23(6): 922-930.
- 51. Luo, W., Wang, Y., Reiser, G. Two types of protease-activated receptors (PAR-1 and PAR-2) mediate calcium signaling in rat retinal ganglion cells RGC-5. (2005) Brain Res 1047(2): 159-167.
- 52. Mann, K.G., Brummel, K., Butenas, S. What is all that thrombin for? (2003) J Thromb Haemost 1(7): 1504-1514.
- 53. Sutherland, C.J., Schurman, J.R. Complications associated with warfarin prophylaxis in total knee arthroplasty. (1987) Clin Orthop Relat Res (219): 158-162.
PubMed||CrossRef||Others
- 54. Goldsack, N.R., Chambers, R.C., Dabbagh, K., et al. Thrombin. (1998) Int J Biochem Cell Biol 30(6): 641-646.
- 55. Haas, S. Future potential indications for an oral thrombin inhibitor. (2002) Hamostaseologie 22(3): 36-43.
PubMed||CrossRef||Others
- 56. Hedner, U. General haemostatic agents--fact or fiction? (2002) Pathophysiol Haemost Thromb 32(Suppl 1): 33-36.
- 57. Mahajan, V.B., Pai, K.S., Lau, A., et al. Creatine kinase, an ATP-generating enzyme, is required for thrombin receptor signaling to the cytoskeleton. (2000) Proc Natl Acad Sci U S A 97(22): 12062-12067.
- 58. Warkentin, T.E., Sikov, W.M., Lillicrap, D.P. Multicentric warfarin-induced skin necrosis complicating heparin-induced thrombocytopenia. (1999) Am J Hematol 62(1): 44-48.
- 59. Fenton, J.W., Ofosu, F.A., Brezniak, D.V., et al. Thrombin and antithrombotics. (1998) Semin Thromb Hemost 24(2): 87-91.
- 60. Stewart, A.J. Penman, I.D., Cook, M.K., et al. Warfarin-induced skin necrosis. (1999) Postgrad Med J 75(882): 233-235.
PubMed||CrossRef||Others
- 61. Brummel, K.E., Paradis, S.G., Butenas, S., et al. Thrombin functions during tissue factor-induced blood coagulation. (2002) Blood 100(1): 148-152.
PubMed||CrossRef||Others
- 62. Chalmers, C.J., Balmanno, K., Hadfield, K., et al. Thrombin inhibits Bim (Bcl-2-interacting mediator of cell death) expression and prevents serum-withdrawal-induced apoptosis via protease-activated receptor 1. (2003) Biochem J 375(Pt 1): 99-109.
- 63. Chinni, C., de Niese, M.R., Tew, D.J., et al. Thrombin, a survival factor for cultured myoblasts. (1999) J Biol Chem 274(14): 9169-9174.
- 64. James, M.F., Roche, A.M. Dose-response relationship between plasma ionized calcium concentration and thrombelastography. (2004) J Cardiothorac Vasc Anesth 18(5): 581-586.
- 65. Shapiro, M.J., Mistry, B. Calcium regulation and nonprotective properties of calcium in surgical ischemia. (1996) New Horiz 4: 134-138.
PubMed||CrossRef||Others
- 66. Bobe, R., Yin, X., Roussanne, M.C., et al. Evidence for ERK1/2 activation by thrombin that is independent of EGFR transactivation. (2003) Am J Physiol Heart Circ Physiol 285(2): H745-754.
- 67. Jeng, J.H., Chan, C.P., Wu, H.L., et al. Protease-activated receptor-1-induced calcium signaling in gingival fibroblasts is mediated by sarcoplasmic reticulum calcium release and extracellular calcium influx. (2004) Cell Signal 16(6): 731-740.
- 68. Brass, E.P., Forman, W.B., Edwards, R.V., et al. Fibrin formation: effect of calcium ions. (1978) Blood 52(4): 654-658.
- 69. Brostrom, M.A., et al. Ca2+ dynamics of thrombin-stimulated rat heart-derived embryonic myocytes: relationship to protein synthesis and cell growth. (2003) Int J Biochem Cell Biol 35(11): 1573-1587.
- 70. Butenas, S., Brummel, K.E., Paradis, S.G., et al. Influence of factor VIIa and phospholipids on coagulation in “acquired” hemophilia. (2003) Arterioscler Thromb Vasc Biol 23(1): 123-129.
- 71. Davies, M.S., Flannery, M.C., McCollum, C.N. Calcium alginate as haemostatic swabs in hip fracture surgery. (1997) J R Coll Surg Edinb 42(1): 31-32.
PubMed||CrossRef||Others
- 72.Gurrieri, M.A. Thrombin-fibrinogen interaction: release of fibrinopeptides and the effects of ATP. (1990) Medicina (Firenze) 10(1): 51-52.
- 73. Murdoch, I.A., Qureshi, S.A., Huggon, I.C. Perioperative haemodynamic effects of an intravenous infusion of calcium chloride in children following cardiac surgery. (1994) Acta Paediatr 83(6): 658-661.
- 74. Sakata, Y., Aoki, N. Cross-linking of alpha 2-plasmin inhibitor to fibrin by fibrin-stabilizing factor. (1980) J Clin Invest 65(2): 290-297.
- 75. von Brecht, J.H., Flanigan, M.J., Freeman, R.M., et al. Regional anticoagulation: hemodialysis with hypertonic trisodium citrate. (1986) Am J Kidney Dis 8(3): 196-201.
PubMed||CrossRef||Others
- 76. Zuccala, G., Pahor, M., Landi, F., et al. Use of calcium antagonists and need for perioperative transfusion in older patients with hip fracture: observational study. (1997) BMJ 314(7081): 643-644.
- 77. Zuccala, G., Pedone, C., Cocchi, A., et al. Use of calcium antagonists and hemoglobin loss in hospitalized elderly patients: a cohort study. Gruppo Italiano di Farmacoepidemiologia nell’Anziano (GIFA) investigators. (2000) Clin Pharmacol Ther 67(3): 314-322.
- 78. Barbagallo, M., Dominguez, L.J., Resnick, L.M. Magnesium metabolism in hypertension and type 2 diabetes mellitus. (2007) Am J Ther 14(4): 375-385.
- 79. , S.B., Worthley, L.I. The essentials of calcium, magnesium and phosphate metabolism: part II. Disorders. (2002) Crit Care Resusc 4(4): 307-315.
- 80. Baker, S.B., Worthley, L.I. The essentials of calcium, magnesium and phosphate metabolism: part I. Physiology. (2002) Crit Care Resusc 4(4): 301-306.
PubMed||CrossRef||Others
- 81. James, M.F., Cronje, L. Pheochromocytoma crisis: the use of magnesium sulfate. (2004) Anesth Analg 99(3): 680-686.
- 82. Dedhia, H.V., Banks, D.E. Pulmonary response to hyperoxia: effects of magnesium. (1994) Environ Health Perspect 102(Suppl 10): 101-105.
PubMed||CrossRef||Others
- 83 Elsharnouby, N.M., Elsharnouby, M.M. Magnesium sulphate as a technique of hypotensive anaesthesia. (2006) Br J Anaesth 96(6): 727-731.
- 84. Elin, R.J. Magnesium metabolism in health and disease. (1988) Dis Mon 34(4): 161-218.
- 85. Erodi, A. Magnesium--an anticoagulant physiological electrolyte. (1973) Med Klin 68: 216-219.
PubMed||CrossRef||Others
- 86. Fehlinger, R., Mielke, U., Fauk, D., et al. Rheographic indications for reduced cerebral vasoconstriction after oral magnesium medication in tetanic patients, a double-blind, placebo-controlled trial. (1986) Magnesium 5(2): 60-65.
PubMed||CrossRef||Others
- 87. Fuentes, A., Rojas, A., Porter, K.B., et al. The effect of magnesium sulfate on bleeding time in pregnancy. (1995) Am J Obstet Gynecol 173(4): 1246-1249.
- 88. Galland, L. Magnesium, stress and neuropsychiatric disorders. (1991) Magnes Trace Elem 10(2-4): 287-301.
PubMed||CrossRef||Others
- 89. Kondo, H., Kobayashi, E., Itani, T., et al. Hematology tests of blood anticoagulated with magnesium sulphate. (2002) Southeast Asian J Trop Med Public Health 33(Suppl 2): 6-9.
PubMed||CrossRef||Others
- 90. Menzel, R., Pusch, H., Ratzmann, G.W., et al. Serum magnesium in insulin-dependent diabetics and healthy subjects in relation to insulin secretion and glycemia during glucose-glucagon test. (1985) Exp Clin Endocrinol 85(1): 81-88.
- 91. Mussoni, L., Sironi, L., Tedeschi, L., et al. Magnesium inhibits arterial thrombi after vascular injury in rat: in vivo impairment of coagulation. (2001) Thromb Haemost 86(5): 1292-1295.
- 92. Rukshin, V., Shah, P.K., Cercek, B., et al. Comparative antithrombotic effects of magnesium sulfate and the platelet glycoprotein IIb/IIIa inhibitors tirofiban and eptifibatide in a canine model of stent thrombosis. (2002) Circulation 105(16): 1970-1975.
- 93. Scheibe, F., Haupt, H., Vlastos, G.A. Preventive magnesium supplement reduces ischemia-induced hearing loss and blood viscosity in the guinea pig. (2000) Eur Arch Otorhinolaryngol 257(7): 355-361.
- 94. Seelig, M.S. Increased need for magnesium with the use of combined oestrogen and calcium for osteoporosis treatment. (1990) Magnes Res 3(3): 197-215.
PubMed||CrossRef||Others
- 95. Pagel, C.N., de Niese, M.R., Abraham, L.A., et al. Inhibition of osteoblast apoptosis by thrombin. (2003) Bone 33(4): 733-743.
- 96. , A., Carney, D.H., Pucci, A., et al. Human alpha-thrombin stimulates proliferation of interferon-gamma differentiated, growth-arrested U937 cells, overcoming differentiation-related changes in expression of p21CIP1/WAF1 and cyclin D1. (2002) J Cell Physiol 191(3): 290-297.
- 97. Brass, L.F., Molino, M. Protease-activated G protein-coupled receptors on human platelets and endothelial cells. (1997) Thromb Haemost 78(1): 234-241.
PubMed||CrossRef||Others
- 98. Bretschneider, E., Kaufmann, R., Braun, M., et al. Evidence for proteinase-activated receptor-2 (PAR-2)-mediated mitogenesis in coronary artery smooth muscle cells. (1999) Br J Pharmacol 126(8): 1735-1740.
- 99. Day, J.R., Taylor, K.M., Lidington, E.A., et al. Aprotinin inhibits proinflammatory activation of endothelial cells by thrombin through the protease-activated receptor 1. (2006) J Thorac Cardiovasc Surg 131(1): 21-27.
- 100. Gorbacheva, L.R., Storozhevykh, T.P., Kiseleva, E.V., et al. Proteinase-activated type 1 receptors are involved in the mechanism of protection of rat hippocampal neurons from glutamate toxicity. (2005) Bull Exp Biol Med 140(3): 285-288.
PubMed||CrossRef||Others
- 101. Wang, K.Y., Chang, F.H., Jeng, J.H., et al. Expression of functional type 1 protease-activated thrombin receptors by mouse primary palatal mesenchymal cells in vitro. (2000) Arch Oral Biol 45(10): 819-825.
- 102. Wang, H., Reiser, G. Thrombin signaling in the brain: the role of protease-activated receptors. (2003) Biol Chem 384(2): 193-202.
- 103. Karp, J.M., Tanaka, T.S., Zohar, R., et al. Thrombin mediated migration of osteogenic cells. (2005) Bone 37(3): 337-348.
- 104. Hong, S.L., Levine, L. Stimulation of prostaglandin synthesis by bradykinin and thrombin and their mechanisms of action on MC5-5 fibroblasts. (1976) J Biol Chem 251(18): 5814-5816.
- 105. Sabri, A., Muske, G., Zhang, H.L., et al. Signaling properties and functions of two distinct cardiomyocyte protease-activated receptors. (2000) Circ Res 86: 1054-1061.
- 106. Pohl, J., Bruhn, H.D., Christophers, E. Thrombin and fibrin-induced growth of fibroblasts: role in wound repair and thrombus organization. (1979) Klin Wochenschr 57(6): 273-277.
- 107. Imamura, T. Tissue factor expression at the site of inflammation: a cross-talk between inflammation and the blood coagulation system. (2004) Rinsho Byori 52: 342-349
PubMed||CrossRef||Others
- 108. Glembotski, C.C., Irons, C.E., Krown, K.A., et al. Myocardial alpha-thrombin receptor activation induces hypertrophy and increases atrial natriuretic factor gene expression. (1993) J Biol Chem 268(27): 20646-20652.
- 109. Okada, M., Suzuki, K., Takada, K., et al. Detection of up-regulated genes in thrombin-stimulated human umbilical vein endothelial cells. (2005) Thromb Res 118(6): 715-721.
- 110. Rothman, A., Wolner, B., Button, D., et al. Immediate-early gene expression in response to hypertrophic and proliferative stimuli in pulmonary arterial smooth muscle cells. (1994) J Biol Chem 269(9): 6399-6404.
- 111. Tsopanoglou, N.E., Maragoudakis, M.E. On the mechanism of thrombin-induced angiogenesis: inhibition of attachment of endothelial cells on basement membrane components. (1998) Angiogenesis 1(2): 192-200.
- 112. Zania, P., Kritikou, S., Flordellis, C.S., et al. Blockade of angiogenesis by small molecule antagonists to protease-activated receptor-1 (PAR-1): Association with endothelial cell growth suppression and induction of apoptosis. (2006) J Pharmacol Exp Ther 318(1): 246-254
- 113. Hornstra, G., Hemker, H.C. Clot-promoting effect of platelet-vessel wall interaction: influence of dietary fats and relation to arterial thrombus formation in rats. (1979) Haemostasis 8(3-5): 211-226.
- 114. Hornstra, G. Platelet - vessel wall interaction: role of blood clotting. (1981) Philos Trans R Soc Lond B Biol Sci 294(1072): 355-371.
- 115. Henriksen, R.A., Samokhin, G.P., Tracy, P.B. Thrombin-induced thromboxane synthesis by human platelets. Properties of anion binding exosite I-independent receptor. (1997) Arterioscler Thromb Vasc Biol 17(12): 3519-3526.
- 116. Tate, B.F., Rittenhouse, S.E. Thrombin activation of human platelets causes tyrosine phosphorylation of PLC-gamma 2. (1993) Biochim Biophys Acta 1178(3): 281-285.
- 117. Garcia, J.G., Pavalko, F.M., Patterson, C.E. Vascular endothelial cell activation and permeability responses to thrombin. (1995) Blood Coagul Fibrinolysis 6(7): 609-626.
- 118. Asero, R., Tedeschi, A., Riboldi, P., et al. Plasma of patients with chronic urticaria shows signs of thrombin generation, and its intradermal injection causes wheal-and-flare reactions much more frequently than autologous serum. (2006) J Allergy Clin Immunol 117(5): 1113-1117.
- 119. Naldini, A., Witkowska – Pelc, E., Filippi, I., et al. Thrombin inhibits IFN-gamma production in human peripheral blood mononuclear cells by promoting a Th2 profile. (2006) J Interferon Cytokine Res 26(11): 793-799.
PubMed||CrossRef||Others
- 120. Naldini, A., Bernini, C., Pucci, A., et al. Thrombin-mediated IL-10 up-regulation involves protease-activated receptor (PAR)-1 expression in human mononuclear leukocytes. (2005) J Leukoc Biol 78(3): 736-744.
- 121. Naldini, A., Carraro, F., Baldari, C.T., et al. The thrombin peptide, TP508, enhances cytokine release and activates signaling events. (2004) Peptides 25(11): 1917-1926.
- 122. Naldini, A., Aarden, L., Pucci, A., et al. Inhibition of interleukin-12 expression by alpha-thrombin in human peripheral blood mononuclear cells: a potential mechanism for modulating Th1/Th2 responses. (2003) Br J Pharmacol 140(5): 980-986.
- 123. Naldini, A., Pucci, A., Carney, D.H., et al. Thrombin enhancement of interleukin-1 expression in mononuclear cells: involvement of proteinase-activated receptor-1. (2002) Cytokine 20(5): 191-199.
- 124. Naldini, A., Carney, D.H., Pucci, A., et al. Thrombin regulates the expression of proangiogenic cytokines via proteolytic activation of protease-activated receptor-1. (2000) Gen Pharmacol 35(5): 255-259.
- 125. Naldini, A., Sower, L., Bocci, V., et al. Thrombin receptor expression and responsiveness of human monocytic cells to thrombin is linked to interferon-induced cellular differentiation. (1998) J Cell Physiol 177(1): 76-84.
- 126. Ben Amor, N., Pariente, J.A., Salido, G.M., et al. Caspases 3 and 9 are translocated to the cytoskeleton and activated by thrombin in human platelets. Evidence for the involvement of PKC and the actin filament polymerization. (2005) Cell Signal 18(8): 1252-1261.
- 127. Ueno, A., Murakami, K., Yamanouchi, K., et al. Thrombin stimulates production of interleukin-8 in human umbilical vein endothelial cells. (1996) Immunology 88(1): 76-81.
- 128. Hua, Y., Keep, R.F., Schallert, T., et al. A thrombin inhibitor reduces brain edema, glioma mass and neurological deficits in a rat glioma model. (2003) Acta Neurochir 86(Suppl): 503-506.
PubMed||CrossRef||Others
- 129. Song, S.J., Pagel, C.N., Campbell, T.M., et al. The role of protease-activated receptor-1 in bone healing. (2005) Am J Pathol 166(3): 857-868.
- 130. Howell, D.C., Goldsack, N.R., Marshall, R.P., et al. Direct thrombin inhibition reduces lung collagen, accumulation, and connective tissue growth factor mRNA levels in bleomycin-induced pulmonary fibrosis. (2001) Am J Pathol 159(4): 1383-1395.
- 131. Howells, G.L., Macey, M., Curtis, M.A., et al. Peripheral blood lymphocytes express the platelet-type thrombin receptor. (1993) Br J Haematol 84(1): 156-160.
- 132. Tordai, A., Fenton, J.W., Andersen, T., et al. Functional thrombin receptors on human T lymphoblastoid cells. (1993) J Immunol 150(11): 4876-4886.
- 133. Naldini, A., Carney, D.H. Thrombin modulation of natural killer activity in human peripheral lymphocytes. (1996) Cell Immunol 172(1): 35-42.
- 134. Naldini, A., Carney, D.H., Bocci, V., et al. Thrombin enhances T cell proliferative responses and cytokine production. (1993) Cell Immunol 147(2): 367-377.
- 135. Huang, Y.Q., Li, J.J., Karpatkin, S. Thrombin inhibits tumor cell growth in association with up-regulation of p21 (waf/cip1) and caspases via a p53-independent, STAT-1-dependent pathway. (2000) J Biol Chem 275(9): 6462-6468.
- Kuznik, B.I., Malezhik, 136. L.P., Al’fonov, V.V., et al. Effect of thrombin on macrophage and lymphocyte functional activity. (1985) Bull Exp Biol Med 99(5): 597-598.
PubMed||CrossRef||Others
- 137. Tran, T., Stewart, A.G. Protease-activated receptor (PAR)-independent growth and pro-inflammatory actions of thrombin on human cultured airway smooth muscle. (2003) Br J Pharmacol 138(5): 865-875.
- 138. Vlahos, R., Lee, K.S., Guida, E., et al. Differential inhibition of thrombin- and EGF-stimulated human cultured airway smooth muscle proliferation by glucocorticoids. (2003) Pulm Pharmacol Ther 16(3): 171-180.
- 139. Laudes, I.J., Chu, J.C., Sikranth, S., et al. Anti-c5a ameliorates coagulation/fibrinolytic protein changes in a rat model of sepsis. (2002) Am J Pathol 160(5): 1867-1875.
- 140. Holm, B., Nilsen, D.W., Kierulf, P., et al. Purification and characterization of 3 fibrinogens with different molecular weights obtained from normal human plasma. (1985) Thromb Res 37(1): 165-176.
- 141. Bouma, B.N., Mosnier, L.O. Thrombin activatable fibrinolysis inhibitor (TAFI)--how does thrombin regulate fibrinolysis? (2006) Ann Med 38(6): 378-388.
- 142. Brass, E.P., Forman, W.B., Edwards, R.V., et al. Fibrin formation: the role of the fibrinogen-fibrin monomer complex. (1976) Thromb Haemost 36(1): 37-48.
PubMed||CrossRef||Others
- 143. Brummel, K.E., Butenas, S., Mann, K.G. An integrated study of fibrinogen during blood coagulation. (1999) J Biol Chem 274(32): 22862-22870.
- 144. Butenas, S., Mann, K.G. Blood coagulation. (2002) Biochemistry (Mosc) 67(1): 3-12.
- 145. Juhan-Vague, I., Hans, M. From fibrinogen to fibrin and its dissolution. (2003) Bull Acad Natl Med 187(1): 69-82; discussion 83-64.
PubMed||CrossRef||Others
- 146. Mosesson, M.W. Fibrinogen and fibrin structure and functions. (2005) J Thromb Haemost 3(8): 1894-1904.
- 147. Malik, A.B. Role of fibrin-neutrophil interactions in lung vascular injury. (1987) Prog Clin Biol Res 236A: 33-42.
PubMed||CrossRef||Others
- 148. Weisel, J.W. Fibrinogen and fibrin. (2005) Adv Protein Chem 70: 247-299.
- 149. Stasko, J., Hudecek, J., Kubisz, P. Thrombin activatable fibrinolysis inhibitor (TAFI) and its importance in the regulation of fibrinolysis. (2004) Vnitr Lek 50(1): 36-44.
PubMed||CrossRef||Others
- 150. Castellino, F.J. Recent Advances in the Chemistry of Fibrinolytic system. (1981) Chem Rev 81: 431-446.
- 151. Castoldi, E., Rosing, J. Factor V Leiden: a disorder of factor V anticoagulant function. (2004) Curr Opin Hematol 11(3): 176-181.
- 152. Walsh, P.N., Ahmad, S.S. Proteases in blood clotting. (2002) Essays Biochem 38: 95-111.
- 153. Smith, D.B., Janmey, P.A., Herbert, T.J., et al. Quantitative measurement of plasma gelsolin and its incorporation into fibrin clots. (1987) J Lab Clin Med 110(2): 189-195.
- 154. Pena, E., Padro, T., Molins, B., et al. Proteomic signature of thrombin-activated platelets after in vivo nitric oxide-donor treatment: coordinated inhibition of signaling (phosphatidylinositol 3-kinase-gamma, 14-3-3zeta, and growth factor receptor-bound protein 2) and cytoskeleton protein translocation. (2011) Arterioscler Thromb Vasc Biol 31(11): 2560-2569.
- 155. Gong, Y., Xi, G.H., Keep, R.F., et al. Complement inhibition attenuates brain edema and neurological deficits induced by thrombin. (2005) Acta Neurochir Suppl 95: 389-392.
- 156. Augustin, H.G., Braun, K., Telemenakis, I., et al. Ovarian angiogenesis. Phenotypic characterization of endothelial cells in a physiological model of blood vessel growth and regression. (1995) Am J Pathol 147(2): 339-351.
- 157. Birkenbach, M., Josefsen, K., Yalamanchili, R., et al. Epstein-Barr virus-induced genes: first lymphocyte-specific G protein-coupled peptide receptors. (1993) J Virol 67(4): 2209-2220.
- 158. Suo, Z., Wu, M., Citron, B.A., et al. Persistent protease-activated receptor 4 signaling mediates thrombin-induced microglial activation. (2003) J Biol Chem 278(33): 31177-31183.
- 159. Zain, J., Huang, Y.Q., Feng, X., et al. Concentration-dependent dual effect of thrombin on impaired growth/apoptosis or mitogenesis in tumor cells. (2000) Blood 95(10): 3133-3138.
- 160., N.E., Maragoudakis, M.E. Role of thrombin in angiogenesis and tumor progression. (2004) Semin Thromb Hemost 30(1): 63-69.
- 161. Nierodzik, M.L., Karpatkin, S. Thrombin induces tumor growth, metastasis, and angiogenesis: Evidence for a thrombin-regulated dormant tumor phenotype. (2006) Cancer cell 10(5): 355-362.
- 162. Choi, S.H., Lee, D.Y., Ryu, J.K., et al. Thrombin induces nigral dopaminergic neurodegeneration in vivo by altering expression of death-related proteins. (2003) Neurobiol Dis 14(2): 181-193.
- 163. Turgeon, V.L., Lloyd, E.D., Wang, S., et al. Thrombin perturbs neurite outgrowth and induces apoptotic cell death in enriched chick spinal motoneuron cultures through caspase activation. (1998) J Neurosci 18(17): 6882-6891.
- 164. Uprichard, J., Perry, D.J. Factor X deficiency. (2002) Blood Rev 16(2): 97-110.
- 165. Idell, S. Endothelium and disordered fibrin turnover in the injured lung: newly recognized pathways. (2002) Crit Care Med 30(5 Suppl): S274-S280.
- 166. Gartner, H.V. Nephropathy in pregnancy--an endothelial lesion? (1994) Zentralbl Gynakol 116: 123-137.
PubMed||CrossRef||Others
- 167. Fan, J., Kapus, A., Li, Y.H., et al. Priming for enhanced alveolar fibrin deposition after hemorrhagic shock: role of tumor necrosis factor. (2000) Am J Respir Cell Mol Biol 22(4): 412-421.
- 168. Akassoglou, K., Akpinar, P., Murray, S., et al. Fibrin is a regulator of Schwann cell migration after sciatic nerve injury in mice. (2003) Neurosci Lett 338(3): 185-188.
- 169. Akassoglou, K., Yu, W.M., Akpinar, P., et al. Fibrin inhibits peripheral nerve remyelination by regulating Schwann cell differentiation. (2002) Neuron 33(6): 861-875.
- 170. Akassoglou, K., Strickland, S. Nervous system pathology: the fibrin perspective. (2002) Biol Chem 383(1): 37-45.
- 171. Akassoglou, K., Kombrinck, K.W., Degen, J.L., et al. Tissue plasminogen activator-mediated fibrinolysis protects against axonal degeneration and demyelination after sciatic nerve injury. (2000) J Cell Biol 149(5): 1157-1166.
- 172. Bredbacka, S., Edner, G. Soluble fibrin and D-dimer as detectors of hypercoagulability in patients with isolated brain trauma. (1994) J Neurosurg Anesthesiol 6(2): 75-82.
- 173. Bredbacka, S., Blomback, M., Wiman, B. Soluble fibrin: a predictor for the development and outcome of multiple organ failure. (1994) Am J Hematol 46(4): 289-294.
- 174. Bredbacka, S., Blomback, M., Hagnevik, K., et al. Per- and postoperative changes in coagulation and fibrinolytic variables during abdominal hysterectomy under epidural or general anaesthesia. (1986) Acta Anaesthesiol Scand 30(3): 204-210.
- 175. Fairley, J.K., Owen, J.E., Birch, D.F. Protein composition of urinary casts from healthy subjects and patients with glomerulonephritis. (1983) Br Med J (Clin Res Ed) 287(6408): 1838-1840.
PubMed||CrossRef||Others
- 176. Ventura, J.E., Villa, M., Mizraji, R., et al. Acute renal failure in pregnancy. (1997) Ren Fail 19(2): 217-220.
PubMed||CrossRef||Others
- 177. Rath, W., Faridi, A., Dudenhausen, J.W. HELLP syndrome. (2000) J Perinat Med 28(4): 249-260.
- 178. Tighe, D., Moss, R., Boghossian, S., et al. Multi-organ damage resulting from experimental faecal peritonitis. (1989) Clin Sci (Lond) 76(3): 269-276.
- 179. Ciavarella, D., Reed, R.L., Counts, R.B., et al. Clotting factor levels and the risk of diffuse microvascular bleeding in the massively transfused patient. (1987) Br J Haematol 67(3): 365-368.
- 180. Goudemand, M. [Plasma fibronectin]. (1983) Rev Fr Transfus Immunohematol 26(3): 279-298.
PubMed||CrossRef||Others
- 181. Kario, K., Matsuo, T. Increased incidence of cardiovascular attacks in the epicenter just after the Hanshin-Awaji earthquake. (1995) Thromb Haemost 74(4): 1207.
- 182. Kario, K., Matsuo, T., Kayaba, K., et al. Earthquake-induced cardiovascular disease and related risk factors in focusing on the Great Hanshin-Awaji Earthquake. (1998) J Epidemiol 8(3): 131-139.
- 183. Kario, K., McEwen, B.S., Pickering, T.G. Disasters and the heart: a review of the effects of earthquake-induced stress on cardiovascular disease. (2003) Hypertens Res 26(5): 355-367.
- 184. Fleck, R.A., Rao, L.V., Rapaport, S.I., et al. Localization of human tissue factor antigen by immunostaining with monospecific, polyclonal anti-human tissue factor antibody. (1990) Thromb Res 59(2): 421-437.
- 185. Nees, S. Coronary flow increases induced by adenosine and adenine nucleotides are mediated by the coronary endothelium: a new principle of the regulation of coronary flow. (1989) Eur Heart J 10(Suppl F): 28-35.
- 186. Federici, A.B., Berkowitz, S.D., Zimmerman, T.S., et al. Proteolysis of von Willebrand factor after thrombolytic therapy in patients with acute myocardial infarction. (1992) Blood 79(1): 38-44.
- 187. Gudmundsdottir, B.R., Marder, V.J., Onundarson, P.T. Risk of excessive bleeding associated with marginally low von Willebrand factor and mild platelet dysfunction. (2007) J Thromb Haemost 5: 274-281.
PubMed||CrossRef||Others
- 188. Hovinga, J.A., Zeerleder, S., Kessler, P., et al. ADAMTS-13, von Willebrand factor and related parameters in severe sepsis and septic shock. (2007) J Thromb Haemost 5(11): 2284-2290.
- 189. Muller, A.M., Skrzynski, C., Skipka, G., et al. Expression of von Willebrand factor by human pulmonary endothelial cells in vivo. (2002) Respiration 69(6): 526-533.
- 190. Sadler, J.E. von Willebrand factor. (1991) J Biol Chem 266: 22777-22780.
PubMed||CrossRef||Others
- 191. Burnstock, G., Ralevic, V. New insights into the local regulation of blood flow by perivascular nerves and endothelium. (1994) Br J Plast Surg 47(8): 527-543.
- 192. Itakura, T., Yamamoto, K., Tohyama, M., et al. Central dual innervation of arterioles and capillaries in the brain. (1977) Stroke 8(3): 360-365.
- 193. Kvasnicka, T. [NO (nitric oxide) and its significance in regulation of vascular homeostasis]. (2003) Vnitr Lek 49(4): 291-296.
PubMed||CrossRef||Others
- 194. Lau, C.S., McLaren, M., Belch, J.J. Factor VIII von Willebrand factor antigen levels correlate with symptom severity in patients with Raynaud’s phenomenon. (1991) Br J Rheumatol 30: 433-436.
- 195. van Zwieten, P.A., Doods, H.N. Muscarinic receptors and drugs in cardiovascular medicine. (1995) Cardiovasc Drugs Ther 9(1): 159-167.
- 196. Y. Henderson, in Cyclopedia of Medicine. (1940).
PubMed||CrossRef||Others
- 197. Vischer, U.M., Wollheim, C.B. Epinephrine induces von Willebrand factor release from cultured endothelial cells: involvement of cyclic AMP-dependent signalling in exocytosis. (1997) Thromb Haemost 77(6): 1182-1188.
PubMed||CrossRef||Others
- 198. Vincent, M.A., Montagnani, M., Quon, M.J. Molecular and physiologic actions of insulin related to production of nitric oxide in vascular endothelium. (2003) Curr Diab Rep 3(4): 279-288.
- 199. Hof, B., van Doorne, C.H., Westerweel, J., et al. Experimental observation of nonlinear traveling waves in turbulent pipe flow. (2004) Science 305(5690): 1594-1598.
- 200. Parker, K.H. Instability in Arterial Blood Flow. N. H. C. H. N. A. Norman, Ed., Cardiovascular Flow Dynamics and Measurements (University Park Press, Chamber of Commerce Building, Baltimore, Md. 21202, Baltimore, Md, 1977).
PubMed||CrossRef||Others
- 201. Adamiec, R., -Chabowska, D., Bednarska, J., et al. Contribution of selected factors of inflammatory creative process in the vascular endothelial damage in the diabetes patients. (2003) Pol Arch Med Wewn 110: 683-689.
PubMed||CrossRef||Others
- 202. Genovese, A., Chiariello, M., Cacciapuoti, A.A., et al. Inhibition of hypoxia-induced cardiac hypertrophy by verapamil in rats. (1980) Basic Res Cardiol 75(6): 757-763.
- 203. Genovese, A., Chiariello, M., Latte, S., et al. Bilateral ventricular hypertrophy in rats exposed to acute or chronic hypobaric hypoxia. (1983) Respiration 44(4): 289-293.
- 204. Genovese, A., Latte, S., Bozzaotre, M., et al. Response of the left ventricular connective tissue to hypoxia. (1983) Res Exp Med (Berl) 183(2): 111-115.
- 205. Genovese, A., De Alfieri, W., Latte, S., et al. Regression of myocardial hypertrophy in the rat following removal of acute or chronic hypobaric hypoxia. (1982) Eur Heart J 3(Suppl A): 161-164.
- 206.Franzoni, F., et al. Plasma antioxidant activity and cutaneous microvascular endothelial function in athletes and sedentary controls. (2004) Biomed Pharmacother 58(8): 432-436.
- 207. Sandercock, G.R., Bromley, P.D., Brodie, D.A. Effects of exercise on heart rate variability: inferences from meta-analysis. (2005) Med Sci Sports Exerc 37(3): 433-439.
- 208. Kario, K., Matsuo, T., Kobayashi, H., et al. Earthquake-induced potentiation of acute risk factors in hypertensive elderly patients: possible triggering of cardiovascular events after a major earthquake. (1997) J Am Coll Cardiol 29(5): 926-933.
- 209. Luostarinen, V., Evers, H., Lyytikainen, M.T., et al. Antithrombotic effects of lidocaine and related compounds on laser-induced microvascular injury. (1981) Acta Anaesthesiol Scand 25(1): 9-11.
- 210. Tobias, M.D., Henry, C., Augostides, Y.G. Lidocaine and bupivacaine exert differential effects on whole blood coagulation. (1999) J Clin Anesth 11(1): 52-55
- 211. Tobias, M.D., Pilla, M.A., Rogers, C., et al. Lidocaine inhibits blood coagulation: implications for epidural blood patch. (1996) Anesth Analg 82(4): 766-769.
- 212. Weksler, B.B., Gillick, M., Pink, J. Effect of propranolol on platelet function. (1977) Blood 49(2): 185-196.
- 213. Chohan, I.S., Singh, I., Vermylen, J., et al. Effect of furosemide on plasma fibrinolytic activity and urokinase excretion. (1977) Exp Hematol 5(2): 153-157.
PubMed||CrossRef||Others
- 214. Khatun, S., et al. Induction of hypercoagulability condition by chronic localized cold stress in rabbits. (1999) Thromb Haemost 81(3): 449-455.
- 215. Francis, C.W., Suchkova, V.N. Ultrasound and thrombolysis. (2001) Vasc Med 6: 181-187.
PubMed||CrossRef||Others
- 216. Suchkova, V.N., Baggs, R.B., Francis, C.W. Effect of 40-kHz ultrasound on acute thrombotic ischemia in a rabbit femoral artery thrombosis model: enhancement of thrombolysis and improvement in capillary muscle perfusion. (2000) Circulation 101(19): 2296-2301.
- 217. Altland, O.D., Dalecki, D., Suchkova, V.N., et al. Low-intensity ultrasound increases endothelial cell nitric oxide synthase activity and nitric oxide synthesis. (2004) J Thromb Haemost 2(4): 637-643.










